Analytical Methods, QA/QC Principles: A Detailed Pharmaceutical Resit
VerifiedAdded on 2024/06/21

PRINCIPLES RESIT
Paraphrase This Document

Table of figures................................................................................................................................3
Q 3(A)...............................................................................................................................................4
Q 3(b)...............................................................................................................................................5
Q 3(c)................................................................................................................................................6
Q 4....................................................................................................................................................9
Q 5 (a)............................................................................................................................................14
Q 5(b).............................................................................................................................................15
References.....................................................................................................................................18

Figure 1: Supercritical Fluid Chromatography.................................................................................5
Figure 2: Conversion method of racemic mixture separation.........................................................6
Figure 3: Thermobalance...............................................................................................................10
Figure 4: Thermogravimetric analysis of CaC2O4.H2O (Heated at the rate of 6°C per minute).....11
Figure 5: Differential Thermal Analysis..........................................................................................12
Figure 6: OOS Investigation...........................................................................................................15
Figure 7: Phase 1 investigation......................................................................................................16
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

DISCUSS WHY PHARMACEUTICAL COMPANIES CARE ABOUT THE CHIRAL PURITY OF THEIR
COMPOUNDS.
The Chiral chromatography helps in no destructive, rapid, and highly selective separation of the
stereoisomers like enantiomers in comparatively less costly. Thus, the use of Chiral Stationary
Phases (CSPs) not only reduces the time of the laboratory but also improves the purity of the
separation. The classical separation techniques used by the laboratories are slow, degradative,
and inefficient as these techniques use chiral resolving reagents before separating the
derivatives. As many of the biochemical pathways are stereoselective, it leads to the selective
crystallization of one stereoisomer only. This method increases the time of the separation
process. In this way, Chirality has become a major approach to synthesize and develop new
drugs (Paithankar, 2013).
The interaction of the drug with biological targets such as bio-membranes, nucleic acids, and
proteins helps in identifying its pharmacological activities. If any disease requires a medicine
with one specific enantiomer the enantiomer doesn't need to have the same impact on the
other disease drug. It can be inactive in another drug or can be toxic. Therefore, it is important
to know the chirality to synthesize stereoisomers effectively for drug preparation (West, 2019).
Reduced laboratory time
Purity improved
Less adverse effects
Enantiomers and other stereoisomers for separation
The application of Supercritical Fluid Chromatography in pharmaceuticals has given a wide
range of advantages to the researchers and analytics. During every stage of drug development,
the active pharmaceutical ingredients and synthesis intermediates can be evaluated for their
enantiopurity. If the observed purity is not sufficient enough then supercritical fluid
chromatography helps in the purification of the desired stereoisomer. This technique not only
Paraphrase This Document

isomer conversion during the process of metabolism which can be used in the detection of the
fate of the drug (Jayakumar et al., 2018).
Figure 1: Supercritical Fluid Chromatography
(Source: West, 2019)
In addition to the influence on the pharmacological activities, bioavailability, safety, and
tolerability, the Stereoselectivity also helps in enabling various drug-drug interactions. In this
way, it helps the researchers to analyze various dimensions of a drug and how it can be useful
for the clinical purpose by assessing the Stereoselectivity of the metabolism of that drug
(Paithankar, 2013).
Q 3(b)
Discuss how racemic mixtures are separated.
The equal proportion of two enantiomers in a mixture that shows no optical rotation of plane-
polarized light is known as a racemic mixture. Such Mixtures can be separated through three
methods into their pure enantiomer elements.

the shape differences, used by Pasteur for the first time. (Serov et al., 2017).
Another method can be the conversion of the enantiomers into diastereomers then
separating it using basic separation techniques from the racemic mixture. In this
method, the diastereomers are treated with reagents that are appropriate to generate
the elementary enantiomers with purity. In the following example, the separation of
original acids is done from the racemic mixture by the addition of HCl solution, and
diastereomers salts are separated by recrystallization
Figure 2: Conversion method of racemic mixture separation
(Source: Serov et al., 2017)
The third method which can be used is the use of Enzymes to separate the molecule.
Enzymes are the catalysts having stereospecific chiral protein properties that enable
them to interact with only one specific enantiomer from the racemic mixture. This
automatically separates the other enantiomer from the mixture and remains unchanged
while the other enantiomer that binds with the enzyme undergoes their specific
reaction. Using an ordinary separation method can be used to separate the unreacted
enantiomer (Serov et al., 2017).
Q 3(c)
If you must validate a specific chromatographic methodology, Discuss how you evaluate the
key validation parameters.
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

preferred analytical methods. It has greater sensitivity, rapidness, ability to reproduce the
result, precision, sample recovery, and high resolution. Validation of an analytical procedure to
determine whether the procedure is suitable for the purpose or not, for which it is intended.
The formulations, discovery, development, and quality control of drugs are based on
chromatographic methods such as HPLC. It helps in assuring the quality of the drugs in the
pharmaceutical industry. In HPLC, the separation of the components depends on the stationary
phase and the extent of its interaction with the solute component. The low-affinity solute will
be eluted first than the higher ones. To valid this process, the coordination of the company with
the quality assurance, analytical, quality control, and regulatory affairs department. A
validation protocol needs to be developed and followed to systematically cover the situation.
The protocol covers the written documentation of the description, policy, structure; individual
responsibility, process description, product description, SOP (standard operating procedure),
resource plan, time plan, controlling plan, and parameters are included. The following are
some key validation parameters that need to be considered (Paithankar, 2013).
Accuracy - The accuracy of this method can be evaluated by the assessment of the
sample against the known concentration. It can be done with the help of using a
certified reference material of known purity. If the quantitation of the impurities needs
to be done, it will require a minimum of nine determinations on the three concentration
levels. These concentration levels should cover the specified range of the sample (Rao,
2018).
Specificity and Selectivity – The specificity is the process that obtains a specific
response to a single analyte. The selectivity refers to the method that quantifies several
analytes, even interferents are present there, whether they are similar or different. Peak
purity or peak homogeneity test is performed to validate the selectivity of the
chromatographic methods like HPLC, as this test has no co-elution of the sample
components. In this test, MS or PDA detectors are used to assess the chromatographic
results (Durão et al., 2017).
Paraphrase This Document

the linearity of the chromatographic method. The regression analysis can be used to
determine the degree of linearity in the sample if it has a linear relationship. The degree
of linearity is shown around the regression line slope (Rao, 2018).
Precision – The repeatability, intermediate precision, and reproducibility are the three
levels of precision based on which the precision of the process can be evaluated.
Repeatability can be at intra-assay intervals in which 6 samples of 100% concentration
are prepared and then assessing the repetition. Intermediate precision can be assessed
using experimental design and testing the samples against standard solutions multiple
times. Reproducibility can be assessed by collaborative studies between different
laboratories (Durão et al., 2017).
Range – The range of the method is also a key parameter of validation which can be
assessed only after the accuracy, linearity, and precision is determined. It depends on
the way procedure is applied; 80-120% of the test concentration for assay method, 70-
130% of the test concentration for content uniformity, near 20% for dissolution, and
from reporting level to 120% of the specified concentration for the impurity
determination (Rao, 2018).

Write an essay critically appraising thermal analysis concerning applications in the
pharmaceutical industry.
Applications of thermal analysis in the Pharmaceutical industry
Thermal analysis has various potential applications in the pharmaceutical sciences as it is an
invaluable technique that helps in characterizing the structure and properties of the
pharmaceutical materials. It is used for the polymorphism investigation the most. The physical
properties that can be tested and determined are loss of weight on drying, decomposition
behavior, polymorphism transition, etc. The thermal degradation, shelf-life, and the
compatibility of excipients require the information of the thermal state of the components that
can be determined by the thermal analysis of the compound. Thus, this kind of analysis is a
valuable insight into the understanding of the kinetics and thermodynamics of the analyte
(Vyazovkin et al., 2018).
The mass, heat capacity, and other properties of the compound are determined following the
temperature that makes it a comparatively easier method. The temperature can be controlled
whether it needs to be kept constant or varied. This way the time, magnitude, and the
temperature of the output of the thermal analysis can be recorded efficiently. The main
methods of thermal analysis are used in the pharmaceutical sector are the Differential thermal
analysis (DTA), Thermogravimetric analysis (TGA), Differential scanning calorimetry (DSC),
Dynamic Mechanical Analysis (DMA), etc. are some of the major thermal analysis techniques
that have been used widely (Qi, 2016).
Thermogravimetric Analysis (TGA)
The Thermogravimetric Analysis (TGA) uses the principle of the change (amount and rate) in the
mass in a controlled environment concerning the rate of change in the temperature. It is used
to identify the physical and chemical processes that took place in the application of heat to it
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

the sample due to heat can be analyzed. In pharmaceutical studies, this method helps in
determining the decomposition, vaporization, and sublimation temperatures. It also helps in
the characterization of the hydrates that originated from the application of the heat to the
sample. To measure and record the thermodynamics of the sample, an instrument called
thermobalance is used, and the graph thus resulted will be called a thermogram.
Thermobalance is consisting of a furnace, autosampler, microgram balance, and a
thermocouple (Qi, 2016).
Figure 3: Thermobalance
(Source - Rao, 2018)
Paraphrase This Document
Thermogravimetric Analysis. Static or Isothermal Thermogravimetric Analysis is the analysis in
which the temperature is kept constant for a particular period and the changes in the mass of
the components of the sample are observed over time. The Dynamic Thermogravimetric
Analysis is the method in which the temperature is increased with control based on
predetermined conditions which result in the invariable linearity with time (Vyazovkin et al.,
2018).
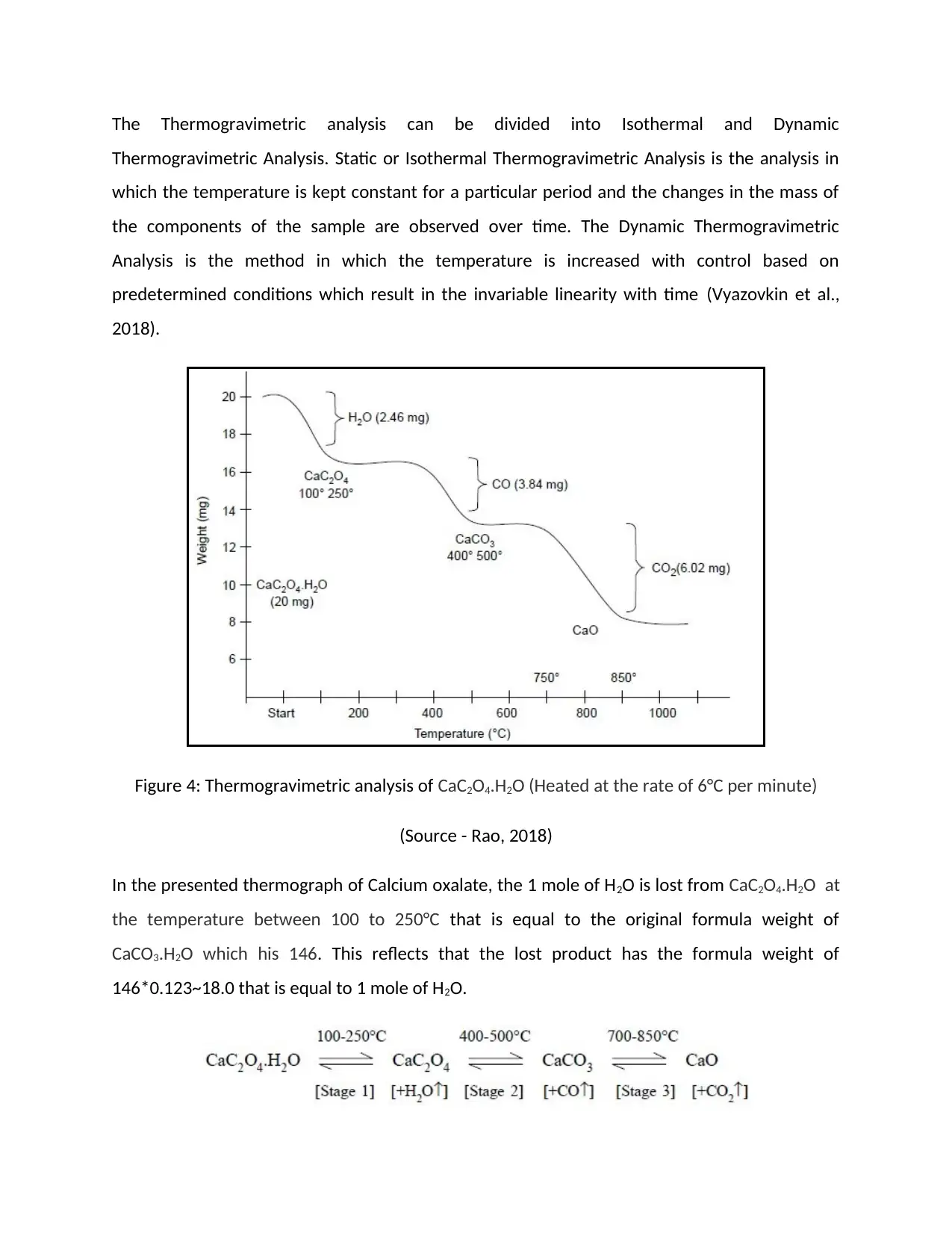
Figure 4: Thermogravimetric analysis of CaC2O4.H2O (Heated at the rate of 6°C per minute)
(Source - Rao, 2018)
In the presented thermograph of Calcium oxalate, the 1 mole of H2O is lost from CaC2O4.H2O at
the temperature between 100 to 250°C that is equal to the original formula weight of
CaCO3.H2O which his 146. This reflects that the lost product has the formula weight of
146*0.123~18.0 that is equal to 1 mole of H2O.
400-500°C that is equivalent to 1 mole of Carbon monoxide occurs. Then, from Calcium
carbonate, the subsequent loss of 3.01% happens between the temperature ranges of 700-
850°C that is equivalent to Carbon-di-oxide. This represents that a compound having multiple
components can be estimated using Thermogravimetric analysis based on their weight
variations at different temperatures. This method can be used for analytical assays, for
screening and testing of analytically substances to form standards for the analytical methods,
and determination of the appropriate forms of elements (Qi, 2016).
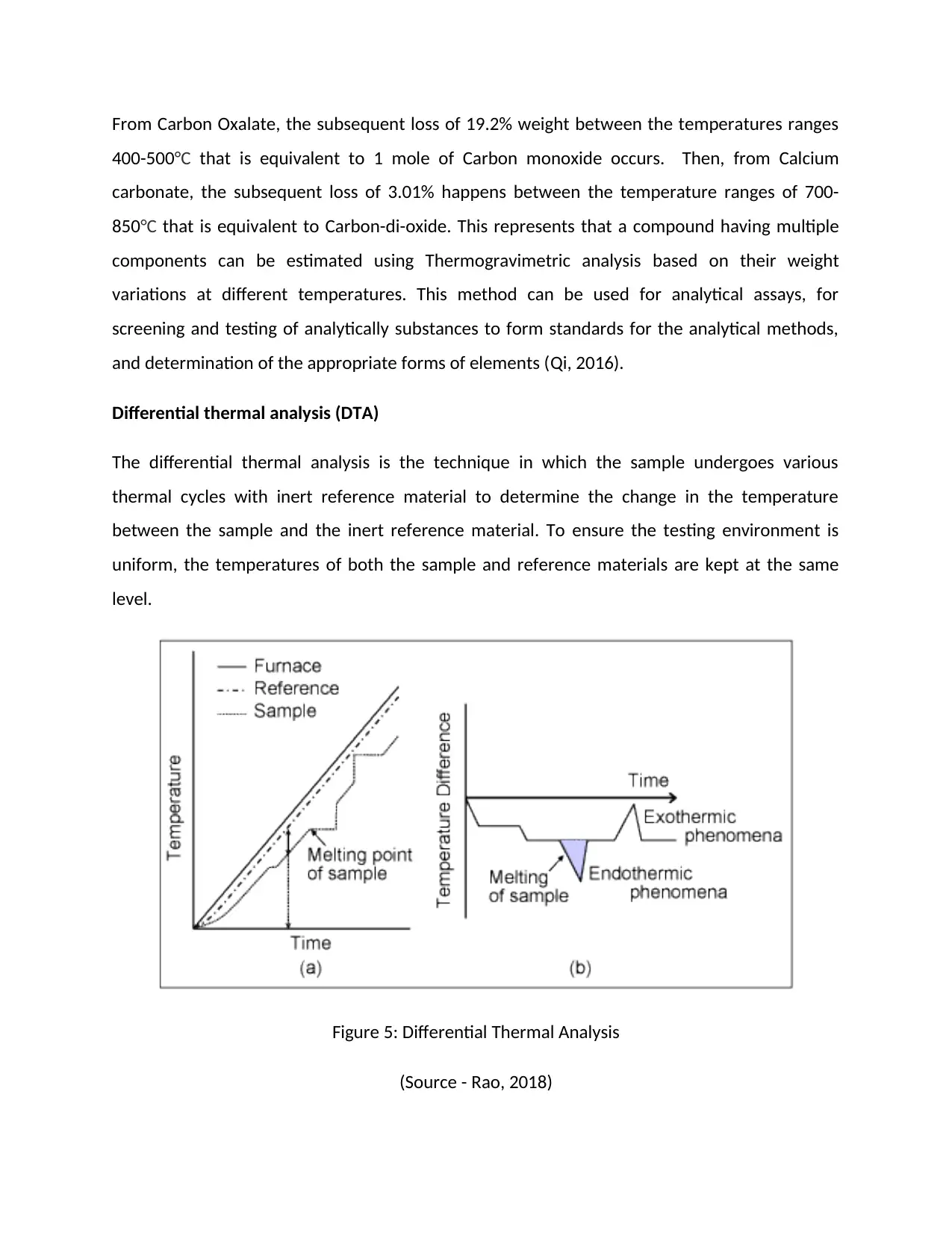
Differential thermal analysis (DTA)
The differential thermal analysis is the technique in which the sample undergoes various
thermal cycles with inert reference material to determine the change in the temperature
between the sample and the inert reference material. To ensure the testing environment is
uniform, the temperatures of both the sample and reference materials are kept at the same
level.
Figure 5: Differential Thermal Analysis
(Source - Rao, 2018)
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

surrounding environment. The thermocouples are used to record the temperatures as the
inbuilt metal wires act as sensors in the thermocouple that can record the temperatures
between cold to hot junctions. The cold junction provides the reference to the hot junction to
measure the temperature of the sample. Voltmeters are also used in this set up to measure the
voltages of the thermocouples and the crucibles used in the furnace to keep the sample. Both
the sample and the reference material are placed symmetrically in the furnace to eliminate the
error of the process (Vyazovkin et al., 2018).
The heating and cooling process is conducted after putting the sample and the reference
material in which the temperatures are tried to keep constant for each cycle of the process. The
recorded differences in the temperature against time are present in a graphical manner which
is called a DTA graph. Reading the graph, the transition temperature of exothermic and
endothermic can be easily determined with the help of the thermocouples as the voltmeter will
read a jump in the temperature every time a phase change occurs (Rao, 2018).
This process can also be used to measure two inert samples to ensure their responses to the
heat cycles. This will help in identifying phase changes without any need for the readings of the
enthalpy change. The information obtained with this technique will help the pharmaceutical
companies in determining the characteristics and impact of the drugs in different conditions.
The polymeric materials can be studied using this technique for their further use in causing
reactions. The heat of reactions, energy change in melting, and specific heat can also be
determined. A drug sample can be tested for its purity and the quality control of several
substances can be done (Vyazovkin et al., 2018).
Paraphrase This Document

What are the key aspects of the analytical method selection process?
Selecting an Analytical method is always a challenging task as the analytical method major part
of the analytical protocol. To select a method, the key aspects that need to be considered are
either generic or specific to a project. It is easier to determine a method when the analytes and
samples are known, and the concentrations are in a small range. To choose the most
appropriate method for the analysis, the requirements of the analysis needs to be determined
first.
Identification of matrix and analyte – The identification of a matrix is done by the analysis of
the characteristics of the necessary sample matrix description. If the description is generic then
the additional information is obtained. The description should include the reason for the in-
homogeneities are due to radioactive particles, sediment particles, another special analyte, or
special chemical forms of the analyte (Kokosa, 2019).
Process knowledge – The operational history and reports by the regulatory can be used for the
process information collection. It should contain sufficient information to decide quickly on the
further planning of the process (Patterson et al., 2016).
Radiological holding and turnaround times – As per MARLAP, the radiological holding time is
the time between the collection and the analysis of the sample. The short half-lives of the
radionuclides should be reviewed to analyze the radiological turnaround time. The selected
method to analyze the analyte should be able to address the constraints of time (Kokosa, 2019).
Unique process specification - The degree of radionuclide heterogeneity in the last sample
prepared at the laboratory, the reporting results of the sample, the weight of dry versus wet
sample, and the length of the sections of sediment core processing are included in the unique
process specification (Kokosa, 2019).

specific method performance should include the specificity, ruggedness, detection capability,
qualification capability, uncertainty, and analyte concentration rage (Patterson et al., 2016).
Q 5(b)
Explain and graphically represent a phase 1a investigation of an OOS result.
The OOS results in the investigation are very useful in the examination of the work that falls
outside the specified criteria. Thus the scope OSS results are based on the criteria that are
accepted in the drug master files and drug applications. The purpose of this investigation is to
find out that the results are relevant and true or not. As the OOS results are generated from the
laboratory, then the investigation of these results needs to be started from the laboratory only.
I any kind of laboratory test, it is crucial that the analysts check whether the results comply with
the test specifications or not. If any kind of unexpected results is found, then the OOS
investigation should be conducted (Kruisz et al., 2017).
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

(Source - Durão et al., 2017)
Phase 1A investigation: The head of the laboratory performs an analysis of the OOS to create
an initial Hypothesis of the investigation. Based on the analysis the Phase 1a investigation is
performed. The main purpose of Phase 1a of the investigation is to ensure whether the OOS
resulted accurately or not. Any kind of deviation in the procedure and practice are determined
and if there are no errors in the testing process the investigation of the OOS results starts.
Paraphrase This Document

(Source: Durão et al., 2017)
There can be errors of incorrect documents or samples in the testing of the sample. The
calculation error or equipment errors can also cause errors in the whole testing. The equipment
can also be showing reading with error due to power outage. The glassware, standards, and
samples can be incorrect that can hugely impact the readings. Based on the issues identified in
the examination the laboratory head decides the root of further investigation. The head will

to be injected again to understand if the problems occur in the performance of the instruments.
The solutions for the test procedure needs to be re-injected to validate that the problems are
arising from the instrumental error, not from the samples or the solutions prepared in the test.
The re-dilution of the solutions from the test can provide evidence that the cause of the
problem is sample or its preparation upon the reinjection of the sample. Once the OOS results
are tested for the errors and there are no significant laboratory errors are found then the
investigation is extended to Phase 1 b. if the result complies with the error of the laboratory in
the testing, then the reports of the investigation are considered for performing the impact and
risk assessment. The individual and mean results are reported to review product history (Kruisz
et al., 2017).
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

Durão, P., Fauteux-Lefebvre, C., Guay, J. M., Abatzoglou, N., and Gosselin, R. (2017).
Using multiple process analytical technology probes to monitor multivitamin blends in a
tableting feed frame. Talanta, 164, 7-15.
Jayakumar, R., Vadivel, R., and Ananthi, N. (2018). Role of chirality in drugs. OMCIJ, 5(3),
1-6.
Kokosa, J. M. (2019). Selecting an extraction solvent for a greener liquid phase
microextraction (LPME) mode-based analytical method. TrAC Trends in Analytical
Chemistry, 118, 238-247.
Kruisz, J., Rehrl, J., Sacher, S., Aigner, I., Horn, M., and Khinast, J. G. (2017). RTD
modeling of a continuous dry granulation process for process control and materials
diversion. International Journal of Pharmaceutics, 528(1-2), 334-344.
Paithankar, H. V. (2013). HPLC method validation for pharmaceuticals: a review. Int J
Universal Pharm Bio Sci, 2(4), 229-240.
Patterson, F., Knight, A., Dowell, J., Nicholson, S., Cousans, F., and Cleland, J. (2016).
How effective are selection methods in medical education? A systematic
review. Medical education, 50(1), 36-60.
Qi, S. (2016). Thermal analysis of pharmaceuticals. In Analytical Techniques in the
Pharmaceutical Sciences (pp. 363-387). Springer, New York, NY.
Rao, T. N. (2018). Validation of Analytical Methods. Calibration and Validation of
Analytical Methods: A Sampling of Current Approaches, 131.
Serov, A., Atanassov, P. B., Kalugin, N., & Frolova, L. V. (2017). U.S. Patent Application
No. 15/515,960.
Shen, Z., Lv, C., and Zeng, S. (2016). Significance and challenges of stereoselectivity
assessing methods in drug metabolism. Journal of pharmaceutical analysis, 6(1), 1-10.
Vyazovkin, S., Koga, N., & Schick, C. (2018). Handbook of thermal analysis and
Calorimetry: Recent Advances, techniques and applications. Elsevier.
Paraphrase This Document

in Analytical Chemistry, 120, 115648.
Your All-in-One AI-Powered Toolkit for Academic Success.
+13062052269
info@desklib.com
Available 24*7 on WhatsApp / Email
![[object Object]](/_next/static/media/star-bottom.7253800d.svg)
© 2024 | Zucol Services PVT LTD | All rights reserved.