Heterotrophic plate count Discussion 2022
VerifiedAdded on 2022/08/24
|10
|9187
|20
AI Summary
Project: Singapore fifth Newater plant Purpose: To enable participants to apply the tools and techniques of decision analysis to a real world problem and, in the process, to deepen their understanding of decision analysis. Assignment question: Identify a public or private sector decision that was reported in the media in the past five years. The problem should be sufficiently “difficult” to merit decision analysis. Taking a standpoint from before the decision was made; your role is to act as consultants to the decision maker(s) by applying the tools of decision analysis to the problem. Your report should include: • Problem statement and objectives >> ~300 - 400 words • Decision tree structure with expected value analysis >> Provide decision tree • Risk profiles >> 350-400 words • Exploration of key uncertainties >>
Contribute Materials
Your contribution can guide someone’s learning journey. Share your
documents today.

Characterization of bacterial community dynamics in a
full-scale drinking water treatment plant
Cuiping Li1, Fangqiong Ling2, Minglu Zhang3, Wen-Tso Liu2, Yuxian Li4, Wenjun Liu1,⁎
1. School of Environment, Tsinghua University, Beijing 100084, China. E-mail: lcp870000@163.com
2. Department of Civil and Environment Engineering, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA
3. School of Food and Chemical Engineering, Beijing Technology and Business University, Beijing 100048, China
4. Water Quality Monitoring Center, Beijing Waterworks Group, Beijing 100085, China
A R T I C L E I N F O A B S T R A C T
Article history:
Received 3 February 2016
Revised 5 May 2016
Accepted 20 May 2016
Available online 2 September 2016
Understanding the spatial and temporal dynamics of microbial communities in drinking
water systems is vital to securing the microbial safety of drinking water. The objective of
this study was to comprehensively characterize the dynamics of microbial biomass and
bacterial communities at each step of a full-scale drinking water treatment plant in Beijing,
China. Both bulk water and biofilm samples on granular activated carbon (GAC) were
collected over 9 months. The proportion of cultivable cells decreased during the treatment
processes, and this proportion was higher in warm season than cool season, suggesting
that treatment processes and water temperature probably had considerable impact on the
R2A cultivability of total bacteria. 16s rRNA gene based 454 pyrosequencing analysis of the
bacterial community revealed that Proteobacteria predominated in all samples. The GAC
biofilm harbored a distinct population with a much higher relative abundance of
Acidobacteria than water samples.Principle coordinate analysis and one-way analysis of
similarity indicated that the dynamics of the microbial communities in bulk water and
biofilm samples were better explained by the treatment processes rather than by sampling
time, and distinctive changes of the microbial communities in water occurred after GAC
filtration. Furthermore, 20 distinct OTUs contributing most to the dissimilarity among
samples of different sampling locations and 6 persistent OTUs present in the entire
treatment process flow were identified. Overall, our findings demonstrate the significant
effects that treatment processes have on the microbial biomass and community fluctuation
and provide implications for further targeted investigation on particular bacteria populations.
© 2016 The Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences.
Published by Elsevier B.V.
Keywords:
Heterotrophic plate count
Flow cytometry
454 pyrosequencing
Bacterial community
Dynamics
Introduction
Drinking water treatment plants (DWTPs) produce potable
water that meets public health regulations from natural water
sources by a series of treatment processes, typically including
coagulation, sedimentation, filtration and disinfection. These
treatment processes result in profound changes in the
physicochemical and biological profiles of the raw water (Au,
2004; Chen et al., 2007). The stable performance of the
treatment processes is crucial to the safety of the treated
water and the microbial communities in drinking water are
particularly important for public health because it is directly
J O U R N A L O F E N V I R O N M E N T A L S C I E N C E S 5 1 ( 2 0 1 7 ) 2 1 – 3 0
⁎ Corresponding author. E-mail: wjliu@tsinghua.edu.cn (Wenjun Liu).
http://dx.doi.org/10.1016/j.jes.2016.05.042
1001-0742/© 2016 The Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences. Published by Elsevier B.V.
A v a i l a b l e o n l i n e a t w w w . s c i e n c e d i r e c t . c o m
ScienceDirect
w w w . e l s e v i e r . c o m / l o c a t e / j e s
full-scale drinking water treatment plant
Cuiping Li1, Fangqiong Ling2, Minglu Zhang3, Wen-Tso Liu2, Yuxian Li4, Wenjun Liu1,⁎
1. School of Environment, Tsinghua University, Beijing 100084, China. E-mail: lcp870000@163.com
2. Department of Civil and Environment Engineering, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA
3. School of Food and Chemical Engineering, Beijing Technology and Business University, Beijing 100048, China
4. Water Quality Monitoring Center, Beijing Waterworks Group, Beijing 100085, China
A R T I C L E I N F O A B S T R A C T
Article history:
Received 3 February 2016
Revised 5 May 2016
Accepted 20 May 2016
Available online 2 September 2016
Understanding the spatial and temporal dynamics of microbial communities in drinking
water systems is vital to securing the microbial safety of drinking water. The objective of
this study was to comprehensively characterize the dynamics of microbial biomass and
bacterial communities at each step of a full-scale drinking water treatment plant in Beijing,
China. Both bulk water and biofilm samples on granular activated carbon (GAC) were
collected over 9 months. The proportion of cultivable cells decreased during the treatment
processes, and this proportion was higher in warm season than cool season, suggesting
that treatment processes and water temperature probably had considerable impact on the
R2A cultivability of total bacteria. 16s rRNA gene based 454 pyrosequencing analysis of the
bacterial community revealed that Proteobacteria predominated in all samples. The GAC
biofilm harbored a distinct population with a much higher relative abundance of
Acidobacteria than water samples.Principle coordinate analysis and one-way analysis of
similarity indicated that the dynamics of the microbial communities in bulk water and
biofilm samples were better explained by the treatment processes rather than by sampling
time, and distinctive changes of the microbial communities in water occurred after GAC
filtration. Furthermore, 20 distinct OTUs contributing most to the dissimilarity among
samples of different sampling locations and 6 persistent OTUs present in the entire
treatment process flow were identified. Overall, our findings demonstrate the significant
effects that treatment processes have on the microbial biomass and community fluctuation
and provide implications for further targeted investigation on particular bacteria populations.
© 2016 The Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences.
Published by Elsevier B.V.
Keywords:
Heterotrophic plate count
Flow cytometry
454 pyrosequencing
Bacterial community
Dynamics
Introduction
Drinking water treatment plants (DWTPs) produce potable
water that meets public health regulations from natural water
sources by a series of treatment processes, typically including
coagulation, sedimentation, filtration and disinfection. These
treatment processes result in profound changes in the
physicochemical and biological profiles of the raw water (Au,
2004; Chen et al., 2007). The stable performance of the
treatment processes is crucial to the safety of the treated
water and the microbial communities in drinking water are
particularly important for public health because it is directly
J O U R N A L O F E N V I R O N M E N T A L S C I E N C E S 5 1 ( 2 0 1 7 ) 2 1 – 3 0
⁎ Corresponding author. E-mail: wjliu@tsinghua.edu.cn (Wenjun Liu).
http://dx.doi.org/10.1016/j.jes.2016.05.042
1001-0742/© 2016 The Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences. Published by Elsevier B.V.
A v a i l a b l e o n l i n e a t w w w . s c i e n c e d i r e c t . c o m
ScienceDirect
w w w . e l s e v i e r . c o m / l o c a t e / j e s
Secure Best Marks with AI Grader
Need help grading? Try our AI Grader for instant feedback on your assignments.

linked to the occurrence of pathogens. As microbial quantity
and composition varied spatially and temporally in drinking
water systems (DWSs) and sometimes the temporal patterns
were striking in both treated and untreated water (Sharp et al.,
2006; Henne et al., 2013; Isaac-Renton et al., 1996), end-product
monitoring alone is inadequate to keep the high level of
confidence in drinking water safety. Holistic characterization
of microbial features in DWTPs is necessary.
Treatment processes such as coagulation, sedimentation
and filtration can physically remove some of the microorgan-
isms. Disinfection inactivates most of the microorganisms,
but those that can endure the disinfection stress are then
transported to distribution networks and proliferated there even
at a low organic nutrient concentration (Boe-Hansen et al., 2002;
Liu et al., 2002;Lu et al., 2014).Various parameters have been
developed to control the biological quality, among which the
total cell count is one of the most widely used parameters
(Carter et al.,2000;Chen et al., 2007;LeChevallier et al.,1991).
Cultivation-based heterotrophic plate count (HPC) method has a
long history to be recommended in most guidelines, but it
detects only a small fraction of the total microbes (Allen et al.,
2004;Bartram et al.,2003).Flow cytometry (FCM) coupled with
nucleic acid targeting stains has been recently developed and
proven to be an sensitive tool for measuring the total cell
concentration in water.The two methods can be integrated to
elucidate the dynamic of cultivable and uncultivable microbial
biomass in DWSs (Hammes et al., 2008).
In terms of community composition, 16S rRNA gene based
sequencing revealed that DWSs harbored diverse microbes,
including some opportunistic pathogens and disinfectant
resistant bacteria in the distribution networks (Pinto et al.,
2012; Berry et al., 2006; Holinger et al., 2014; Hwang et al., 2012;
Lautenschlager et al., 2014; Lin et al., 2014; Mi et al., 2015). The
microorganisms that survived after water treatment process-
es are considered to be an important source for the potential
pathogens at tap faucets. Moreover, the seeded bacteria in
some bio-enhanced filters that used to remove specialized
pollutants posed a potential risk of leaking from filter biofilms
(Zhang et al., 2013).
Some studies concurred that filtration and disinfection
had more significant effects on the microbial community
compared with other processes, such as coagulation and
sedimentation, and that the biofiltration process may deter-
mine the characteristics of the downstream microbiome
(Holinger et al., 2014; Kwon et al., 2011; Pinto et al., 2012;
Wang et al., 2013). Meanwhile, another study showed no
major changes occurred after sand filtration (Eichler et al.,
2006). The discrepancies found among studies may result
from different factors such as study areas, treatment process
chains, time scale of sampling and sequencing methods. The
temporal fluctuation is a critical consideration for rigorous
statistical tests and high validity of information on the
processes. However, longitudinal surveys on the dynamics of
microbial communities through treatment processes were
still limited.
In the present study, the temporal and spatial changes in
microbial communities of a full-scale drinking water treat-
ment plant (DWTP) in Beijing, China, were investigated over
9 months. HPC, FCM and 454 pyrosequencing were used to
measure the microbial biomass and bacterial compositions of
both bulk water and granular activated carbon (GAC) biofilms.
An in-depth characterization of the microbial dynamic
patterns in a DWTP was conducted, the results of this study
may help to extend our knowledge about the microbial quality
of water in DWTPs. The primary objective of this work was
to determine (1) how the treatment processes and the temporal
variation contribute to the microbial biomass and the bacterial
community structure, (2) the influence of GAC biofilm on the
water microbiology and (3) the distinct and persistent bacteria
that present throughout the treatment processes.
1. Materials and methods
1.1. Drinking water treatment processes and sampling schedule
The DWTP monitored in this study produces 60% of the
drinking water of Beijing, China. During the sampling period,
its water source consists of two reservoirs (Miyun reservoir in
Beijing and Huangbizhuang reservoir in Hebei) as well as
groundwater from Huairou aquifer. The average volume
mix ratio of these water sources is 4:1:2. The treatment
processes include pre-chlorination, coagulation, clarification
and coal-sand dual media filtration as the conventional treat-
ment and GAC filtration as the advanced treatment (Fig. 1). The
GAC tanks are backwashed every 6 days. Free chlorine is added
to the GAC effluent at a concentration of 1.2–1.8 mg/L for 5 hr.
0.2 mg/L ammonia is added post clear well to produce a
chloramine residual of 0.7–0.8 mg/L in the distribution system.
Samples were collected in 6 months over a period of
9 months in 2012 (May, August, October, November and
December) and 2013 (January).The pre-chlorinated raw water
(RW), the coal-sand filter effluent (SE),the GAC tank effluent
(CE) and the finished water (FW) were water samples and the
GAC particles (CB) were biofilm samples. The GAC particles
were taken from the top of the filter tank. Samples were
collected in sterile bottles, which were taken to the laboratory
within 4 hr. Water quality parameters were listed in Appendix
A Table S1.
1.2. Heterotrophic plate count
1-mL aliquots of ten-fold serial dilutions of each water sample
were mixed with 20 mL R2A agar (Difco, BD, USA) and
incubated at 20°C for 7 days. All HPC determinations were
performed in triplicate.
1.3. Total cell concentrations measured by flow cytometry
Total cell concentrations were determined according to the
method introduced by Hammes et al. (2008) with a Cell Lab
Quanta SC flow cytometer (Beckman Coulter, Inc., Brea, CA,
USA). The total cell concentration of all the samples should be
maintained between 3 × 103 and 2 × 106 cells/mL.
1.4. DNA extraction
10–40 L of bulk water were filtered through a 0.22-μm pore size
membrane (47 mm diameter, Millipore, USA) with a 90-mm
Filter Holder (Millipore, USA). For GAC biofilm samples, about
22 J O U R N A L O F E N V I R O N M E N T A L S C I E N C E S 5 1 ( 2 0 1 7 ) 2 1 – 3 0
and composition varied spatially and temporally in drinking
water systems (DWSs) and sometimes the temporal patterns
were striking in both treated and untreated water (Sharp et al.,
2006; Henne et al., 2013; Isaac-Renton et al., 1996), end-product
monitoring alone is inadequate to keep the high level of
confidence in drinking water safety. Holistic characterization
of microbial features in DWTPs is necessary.
Treatment processes such as coagulation, sedimentation
and filtration can physically remove some of the microorgan-
isms. Disinfection inactivates most of the microorganisms,
but those that can endure the disinfection stress are then
transported to distribution networks and proliferated there even
at a low organic nutrient concentration (Boe-Hansen et al., 2002;
Liu et al., 2002;Lu et al., 2014).Various parameters have been
developed to control the biological quality, among which the
total cell count is one of the most widely used parameters
(Carter et al.,2000;Chen et al., 2007;LeChevallier et al.,1991).
Cultivation-based heterotrophic plate count (HPC) method has a
long history to be recommended in most guidelines, but it
detects only a small fraction of the total microbes (Allen et al.,
2004;Bartram et al.,2003).Flow cytometry (FCM) coupled with
nucleic acid targeting stains has been recently developed and
proven to be an sensitive tool for measuring the total cell
concentration in water.The two methods can be integrated to
elucidate the dynamic of cultivable and uncultivable microbial
biomass in DWSs (Hammes et al., 2008).
In terms of community composition, 16S rRNA gene based
sequencing revealed that DWSs harbored diverse microbes,
including some opportunistic pathogens and disinfectant
resistant bacteria in the distribution networks (Pinto et al.,
2012; Berry et al., 2006; Holinger et al., 2014; Hwang et al., 2012;
Lautenschlager et al., 2014; Lin et al., 2014; Mi et al., 2015). The
microorganisms that survived after water treatment process-
es are considered to be an important source for the potential
pathogens at tap faucets. Moreover, the seeded bacteria in
some bio-enhanced filters that used to remove specialized
pollutants posed a potential risk of leaking from filter biofilms
(Zhang et al., 2013).
Some studies concurred that filtration and disinfection
had more significant effects on the microbial community
compared with other processes, such as coagulation and
sedimentation, and that the biofiltration process may deter-
mine the characteristics of the downstream microbiome
(Holinger et al., 2014; Kwon et al., 2011; Pinto et al., 2012;
Wang et al., 2013). Meanwhile, another study showed no
major changes occurred after sand filtration (Eichler et al.,
2006). The discrepancies found among studies may result
from different factors such as study areas, treatment process
chains, time scale of sampling and sequencing methods. The
temporal fluctuation is a critical consideration for rigorous
statistical tests and high validity of information on the
processes. However, longitudinal surveys on the dynamics of
microbial communities through treatment processes were
still limited.
In the present study, the temporal and spatial changes in
microbial communities of a full-scale drinking water treat-
ment plant (DWTP) in Beijing, China, were investigated over
9 months. HPC, FCM and 454 pyrosequencing were used to
measure the microbial biomass and bacterial compositions of
both bulk water and granular activated carbon (GAC) biofilms.
An in-depth characterization of the microbial dynamic
patterns in a DWTP was conducted, the results of this study
may help to extend our knowledge about the microbial quality
of water in DWTPs. The primary objective of this work was
to determine (1) how the treatment processes and the temporal
variation contribute to the microbial biomass and the bacterial
community structure, (2) the influence of GAC biofilm on the
water microbiology and (3) the distinct and persistent bacteria
that present throughout the treatment processes.
1. Materials and methods
1.1. Drinking water treatment processes and sampling schedule
The DWTP monitored in this study produces 60% of the
drinking water of Beijing, China. During the sampling period,
its water source consists of two reservoirs (Miyun reservoir in
Beijing and Huangbizhuang reservoir in Hebei) as well as
groundwater from Huairou aquifer. The average volume
mix ratio of these water sources is 4:1:2. The treatment
processes include pre-chlorination, coagulation, clarification
and coal-sand dual media filtration as the conventional treat-
ment and GAC filtration as the advanced treatment (Fig. 1). The
GAC tanks are backwashed every 6 days. Free chlorine is added
to the GAC effluent at a concentration of 1.2–1.8 mg/L for 5 hr.
0.2 mg/L ammonia is added post clear well to produce a
chloramine residual of 0.7–0.8 mg/L in the distribution system.
Samples were collected in 6 months over a period of
9 months in 2012 (May, August, October, November and
December) and 2013 (January).The pre-chlorinated raw water
(RW), the coal-sand filter effluent (SE),the GAC tank effluent
(CE) and the finished water (FW) were water samples and the
GAC particles (CB) were biofilm samples. The GAC particles
were taken from the top of the filter tank. Samples were
collected in sterile bottles, which were taken to the laboratory
within 4 hr. Water quality parameters were listed in Appendix
A Table S1.
1.2. Heterotrophic plate count
1-mL aliquots of ten-fold serial dilutions of each water sample
were mixed with 20 mL R2A agar (Difco, BD, USA) and
incubated at 20°C for 7 days. All HPC determinations were
performed in triplicate.
1.3. Total cell concentrations measured by flow cytometry
Total cell concentrations were determined according to the
method introduced by Hammes et al. (2008) with a Cell Lab
Quanta SC flow cytometer (Beckman Coulter, Inc., Brea, CA,
USA). The total cell concentration of all the samples should be
maintained between 3 × 103 and 2 × 106 cells/mL.
1.4. DNA extraction
10–40 L of bulk water were filtered through a 0.22-μm pore size
membrane (47 mm diameter, Millipore, USA) with a 90-mm
Filter Holder (Millipore, USA). For GAC biofilm samples, about
22 J O U R N A L O F E N V I R O N M E N T A L S C I E N C E S 5 1 ( 2 0 1 7 ) 2 1 – 3 0

15 g wet carbon particles were rinsed twice with sterile PBS
buffer to remove planktonic cells and then immersed in 30 mL
sterile PBS buffer. Ultrasonication (500 W, 40 kHz, 20 min)
was applied to detach bacteria from carbon particles combin-
ing vortexing or hand-shaking for 30 sec every 10 min. Ice
bags were used to prevent the thermal effect caused by
ultrasonication. After ultrasonication, the suspension was
transferred and centrifuged at 10,000 ×g for 10 min at 4°C to
collect the pellet of microbes and the supernatant was
discarded.
The total DNA was extracted from the filter membranes
and the pellets of GAC biofilm with an EZNA® Soil DNA kit
(Omega Bio-Tek, USA) according to the manufacturer's protocol.
The concentration and purity of the total DNA were measured
by a NanoDrop 2000 spectrophotometry (Thermo Fisher Scien-
tific, Wilmington, DE, USA).
1.5. 454 pyrosequencing and data analysis
The hypervariable V1-V3 region of the 16S rRNA gene was
amplified using the universal bacterial primers 27F (5′-AGA
GTTTGATCCTGGCTCAG-3′) and 533R (5′-TTACCGCGGCTGC
TGGCAC-3′). Pyrosequencing was performed on the Roche
454 FLX pyrosequencing platform at SinoGenoMax (Beijing,
China). Pyrosequencing results were processed using the
Quantitative Insights Into Microbial Ecology (QIIME) (v 1.5.0)
pipeline (Caporaso et al., 2010).Briefly, raw sequences were
first filtered with a minimum average quality score ≥ 25, a
sliding window value of 50 and size between 200 and 700 bp;
then denoising was applied to reduce erroneous sequences
with imprecise signals. After denoising, the Uclust algorithm
was used for operational taxonomic unit (OTU) picking (97%
similarity). Representative sequences of each OTU were
selected for alignment to the Greengenes aligned reference
set by PyNAST with default settings. Chimera Slayer was used
to identify chimeras, which were then removed from the
representative sequences. Taxonomy assignment was per-
formed with the RDP classifier using the Greengenes reference
at a 0.8 minimum confidence level. Alpha and beta diversities
were calculated at a sequencing depth of 950 after removing
the singleton sequences. Principal coordinate analysis (PCoA)
was conducted with QIIME to visualize pairwise Bray–Curtis
distance among samples.
Analysis of Similarity (ANOSIM) and Similarity Percentage
(SIMPER) was performed based on the Bray–Curtis dissimilar-
ity distance matrices at OTU level with the software PAST.
ANOSIM is a non-parametric test of significant difference
between two or more groups based on any distance measure.
Distances are converted into ranks prior to calculating the
statistic R. R was calculated by Eq. (1):
R ¼ rb−rw
1
4n n−1ð Þ
ð1Þ
where, rb is the mean rank of all distances between groups, rw
is the mean rank of all distances within the same group, and n
is the total number of samples (Clarke, 1993). R ranges from −1
to 1 and larger R indicates higher degree of separation
between groups. The significance is computed by random
permutation of group membership. SIMPER is used to weight
the contribution of each OTU to the dissimilarity between
groups confirmed to be significant by ANOSIM.
Venn diagram was plotted to identify the numbers of
shared OTUs among different sampling locations with the R
package. OTUs that were present in at least 50% of samples at
each sampling location were picked out respectively and they
were the source data for the Venn diagram.
2. Results and discussion
2.1. Planktonic biomass changes through treatment processes
Microbial biomass in raw water based on HPC and total cell
concentration (FCM) ranged from 1.4 × 103 to 5.9 × 104 CFU/mL
and 2.7 × 105 to 3.9 × 105 cells/mL, respectively (Fig. 2). The
biomass was significantly higher in May, August and October
2012 than in other months, and positive correlation was
observed between the biomass and the water temperature
(Appendix A Table S2) (Pearson's R = 0.841 for HPC and 0.877 for
total cell concentration). Conventional treatment processes
had an average removal rate of 61.6% for HPC and 37.7% for
total cell concentration, whereas the average removal rates of
GAC filtration were 32.1% and 8.3% for HPC and total cell
concentration, respectively. No cultivable bacteria (HPCs) were
detected in all the finished water samples, which met the
Raw
water
Coagulants
(PAC, FeCl3)
Distribution
system
Mechanical
acceleration
clarifying tank
Coal-sand
filter
Mixing
well
Pumping
station
Clear
well
Granular
activated
carbon tank
RW SE
CEFW
CB
ChlorineAmmonia
Fig. 1 – Schematic diagram of the drinking water treatment process. RW: raw water; SE: coal–sand filter effluent; CB: GAC
particles; CE: GAC tank effluent; FW: finished water; GAC: granular activated carbon.
23J O U R N A L O F E N V I R O N M E N T A L S C I E N C E S 5 1 ( 2 0 1 7 ) 2 1 – 3 0
buffer to remove planktonic cells and then immersed in 30 mL
sterile PBS buffer. Ultrasonication (500 W, 40 kHz, 20 min)
was applied to detach bacteria from carbon particles combin-
ing vortexing or hand-shaking for 30 sec every 10 min. Ice
bags were used to prevent the thermal effect caused by
ultrasonication. After ultrasonication, the suspension was
transferred and centrifuged at 10,000 ×g for 10 min at 4°C to
collect the pellet of microbes and the supernatant was
discarded.
The total DNA was extracted from the filter membranes
and the pellets of GAC biofilm with an EZNA® Soil DNA kit
(Omega Bio-Tek, USA) according to the manufacturer's protocol.
The concentration and purity of the total DNA were measured
by a NanoDrop 2000 spectrophotometry (Thermo Fisher Scien-
tific, Wilmington, DE, USA).
1.5. 454 pyrosequencing and data analysis
The hypervariable V1-V3 region of the 16S rRNA gene was
amplified using the universal bacterial primers 27F (5′-AGA
GTTTGATCCTGGCTCAG-3′) and 533R (5′-TTACCGCGGCTGC
TGGCAC-3′). Pyrosequencing was performed on the Roche
454 FLX pyrosequencing platform at SinoGenoMax (Beijing,
China). Pyrosequencing results were processed using the
Quantitative Insights Into Microbial Ecology (QIIME) (v 1.5.0)
pipeline (Caporaso et al., 2010).Briefly, raw sequences were
first filtered with a minimum average quality score ≥ 25, a
sliding window value of 50 and size between 200 and 700 bp;
then denoising was applied to reduce erroneous sequences
with imprecise signals. After denoising, the Uclust algorithm
was used for operational taxonomic unit (OTU) picking (97%
similarity). Representative sequences of each OTU were
selected for alignment to the Greengenes aligned reference
set by PyNAST with default settings. Chimera Slayer was used
to identify chimeras, which were then removed from the
representative sequences. Taxonomy assignment was per-
formed with the RDP classifier using the Greengenes reference
at a 0.8 minimum confidence level. Alpha and beta diversities
were calculated at a sequencing depth of 950 after removing
the singleton sequences. Principal coordinate analysis (PCoA)
was conducted with QIIME to visualize pairwise Bray–Curtis
distance among samples.
Analysis of Similarity (ANOSIM) and Similarity Percentage
(SIMPER) was performed based on the Bray–Curtis dissimilar-
ity distance matrices at OTU level with the software PAST.
ANOSIM is a non-parametric test of significant difference
between two or more groups based on any distance measure.
Distances are converted into ranks prior to calculating the
statistic R. R was calculated by Eq. (1):
R ¼ rb−rw
1
4n n−1ð Þ
ð1Þ
where, rb is the mean rank of all distances between groups, rw
is the mean rank of all distances within the same group, and n
is the total number of samples (Clarke, 1993). R ranges from −1
to 1 and larger R indicates higher degree of separation
between groups. The significance is computed by random
permutation of group membership. SIMPER is used to weight
the contribution of each OTU to the dissimilarity between
groups confirmed to be significant by ANOSIM.
Venn diagram was plotted to identify the numbers of
shared OTUs among different sampling locations with the R
package. OTUs that were present in at least 50% of samples at
each sampling location were picked out respectively and they
were the source data for the Venn diagram.
2. Results and discussion
2.1. Planktonic biomass changes through treatment processes
Microbial biomass in raw water based on HPC and total cell
concentration (FCM) ranged from 1.4 × 103 to 5.9 × 104 CFU/mL
and 2.7 × 105 to 3.9 × 105 cells/mL, respectively (Fig. 2). The
biomass was significantly higher in May, August and October
2012 than in other months, and positive correlation was
observed between the biomass and the water temperature
(Appendix A Table S2) (Pearson's R = 0.841 for HPC and 0.877 for
total cell concentration). Conventional treatment processes
had an average removal rate of 61.6% for HPC and 37.7% for
total cell concentration, whereas the average removal rates of
GAC filtration were 32.1% and 8.3% for HPC and total cell
concentration, respectively. No cultivable bacteria (HPCs) were
detected in all the finished water samples, which met the
Raw
water
Coagulants
(PAC, FeCl3)
Distribution
system
Mechanical
acceleration
clarifying tank
Coal-sand
filter
Mixing
well
Pumping
station
Clear
well
Granular
activated
carbon tank
RW SE
CEFW
CB
ChlorineAmmonia
Fig. 1 – Schematic diagram of the drinking water treatment process. RW: raw water; SE: coal–sand filter effluent; CB: GAC
particles; CE: GAC tank effluent; FW: finished water; GAC: granular activated carbon.
23J O U R N A L O F E N V I R O N M E N T A L S C I E N C E S 5 1 ( 2 0 1 7 ) 2 1 – 3 0
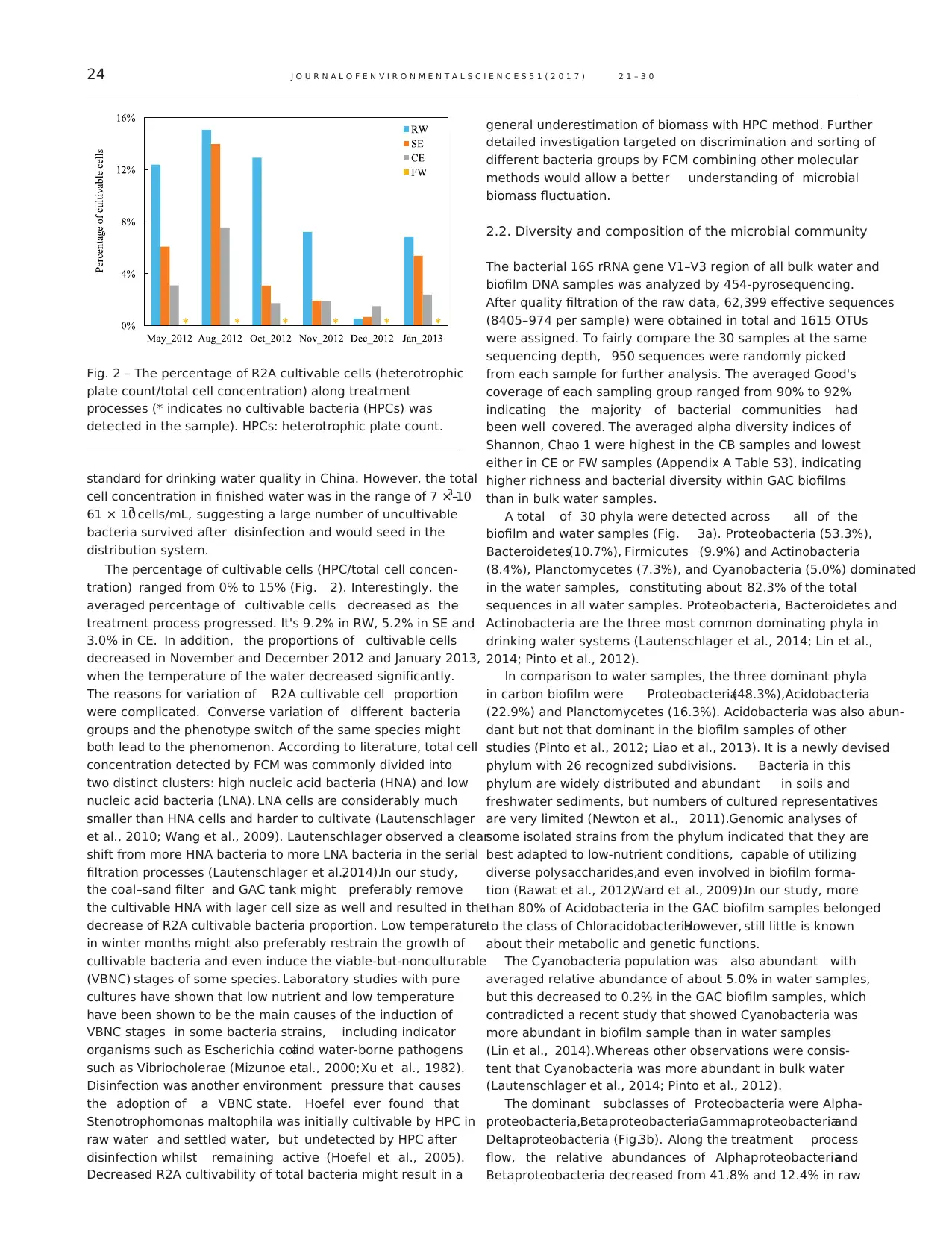
standard for drinking water quality in China. However, the total
cell concentration in finished water was in the range of 7 × 103–
61 × 103 cells/mL, suggesting a large number of uncultivable
bacteria survived after disinfection and would seed in the
distribution system.
The percentage of cultivable cells (HPC/total cell concen-
tration) ranged from 0% to 15% (Fig. 2). Interestingly, the
averaged percentage of cultivable cells decreased as the
treatment process progressed. It's 9.2% in RW, 5.2% in SE and
3.0% in CE. In addition, the proportions of cultivable cells
decreased in November and December 2012 and January 2013,
when the temperature of the water decreased significantly.
The reasons for variation of R2A cultivable cell proportion
were complicated. Converse variation of different bacteria
groups and the phenotype switch of the same species might
both lead to the phenomenon. According to literature, total cell
concentration detected by FCM was commonly divided into
two distinct clusters: high nucleic acid bacteria (HNA) and low
nucleic acid bacteria (LNA). LNA cells are considerably much
smaller than HNA cells and harder to cultivate (Lautenschlager
et al., 2010; Wang et al., 2009). Lautenschlager observed a clear
shift from more HNA bacteria to more LNA bacteria in the serial
filtration processes (Lautenschlager et al.,2014).In our study,
the coal–sand filter and GAC tank might preferably remove
the cultivable HNA with lager cell size as well and resulted in the
decrease of R2A cultivable bacteria proportion. Low temperature
in winter months might also preferably restrain the growth of
cultivable bacteria and even induce the viable-but-nonculturable
(VBNC) stages of some species. Laboratory studies with pure
cultures have shown that low nutrient and low temperature
have been shown to be the main causes of the induction of
VBNC stages in some bacteria strains, including indicator
organisms such as Escherichia coliand water-borne pathogens
such as Vibriocholerae (Mizunoe etal., 2000;Xu et al., 1982).
Disinfection was another environment pressure that causes
the adoption of a VBNC state. Hoefel ever found that
Stenotrophomonas maltophila was initially cultivable by HPC in
raw water and settled water, but undetected by HPC after
disinfection whilst remaining active (Hoefel et al., 2005).
Decreased R2A cultivability of total bacteria might result in a
general underestimation of biomass with HPC method. Further
detailed investigation targeted on discrimination and sorting of
different bacteria groups by FCM combining other molecular
methods would allow a better understanding of microbial
biomass fluctuation.
2.2. Diversity and composition of the microbial community
The bacterial 16S rRNA gene V1–V3 region of all bulk water and
biofilm DNA samples was analyzed by 454-pyrosequencing.
After quality filtration of the raw data, 62,399 effective sequences
(8405–974 per sample) were obtained in total and 1615 OTUs
were assigned. To fairly compare the 30 samples at the same
sequencing depth, 950 sequences were randomly picked
from each sample for further analysis. The averaged Good's
coverage of each sampling group ranged from 90% to 92%
indicating the majority of bacterial communities had
been well covered. The averaged alpha diversity indices of
Shannon, Chao 1 were highest in the CB samples and lowest
either in CE or FW samples (Appendix A Table S3), indicating
higher richness and bacterial diversity within GAC biofilms
than in bulk water samples.
A total of 30 phyla were detected across all of the
biofilm and water samples (Fig. 3a). Proteobacteria (53.3%),
Bacteroidetes(10.7%), Firmicutes (9.9%) and Actinobacteria
(8.4%), Planctomycetes (7.3%), and Cyanobacteria (5.0%) dominated
in the water samples, constituting about 82.3% of the total
sequences in all water samples. Proteobacteria, Bacteroidetes and
Actinobacteria are the three most common dominating phyla in
drinking water systems (Lautenschlager et al., 2014; Lin et al.,
2014; Pinto et al., 2012).
In comparison to water samples, the three dominant phyla
in carbon biofilm were Proteobacteria(48.3%),Acidobacteria
(22.9%) and Planctomycetes (16.3%). Acidobacteria was also abun-
dant but not that dominant in the biofilm samples of other
studies (Pinto et al., 2012; Liao et al., 2013). It is a newly devised
phylum with 26 recognized subdivisions. Bacteria in this
phylum are widely distributed and abundant in soils and
freshwater sediments, but numbers of cultured representatives
are very limited (Newton et al., 2011).Genomic analyses of
some isolated strains from the phylum indicated that they are
best adapted to low-nutrient conditions, capable of utilizing
diverse polysaccharides,and even involved in biofilm forma-
tion (Rawat et al., 2012;Ward et al., 2009).In our study, more
than 80% of Acidobacteria in the GAC biofilm samples belonged
to the class of Chloracidobacteria.However, still little is known
about their metabolic and genetic functions.
The Cyanobacteria population was also abundant with
averaged relative abundance of about 5.0% in water samples,
but this decreased to 0.2% in the GAC biofilm samples, which
contradicted a recent study that showed Cyanobacteria was
more abundant in biofilm sample than in water samples
(Lin et al., 2014).Whereas other observations were consis-
tent that Cyanobacteria was more abundant in bulk water
(Lautenschlager et al., 2014; Pinto et al., 2012).
The dominant subclasses of Proteobacteria were Alpha-
proteobacteria,Betaproteobacteria,Gammaproteobacteriaand
Deltaproteobacteria (Fig.3b). Along the treatment process
flow, the relative abundances of Alphaproteobacteriaand
Betaproteobacteria decreased from 41.8% and 12.4% in raw
Fig. 2 – The percentage of R2A cultivable cells (heterotrophic
plate count/total cell concentration) along treatment
processes (* indicates no cultivable bacteria (HPCs) was
detected in the sample). HPCs: heterotrophic plate count.
24 J O U R N A L O F E N V I R O N M E N T A L S C I E N C E S 5 1 ( 2 0 1 7 ) 2 1 – 3 0
cell concentration in finished water was in the range of 7 × 103–
61 × 103 cells/mL, suggesting a large number of uncultivable
bacteria survived after disinfection and would seed in the
distribution system.
The percentage of cultivable cells (HPC/total cell concen-
tration) ranged from 0% to 15% (Fig. 2). Interestingly, the
averaged percentage of cultivable cells decreased as the
treatment process progressed. It's 9.2% in RW, 5.2% in SE and
3.0% in CE. In addition, the proportions of cultivable cells
decreased in November and December 2012 and January 2013,
when the temperature of the water decreased significantly.
The reasons for variation of R2A cultivable cell proportion
were complicated. Converse variation of different bacteria
groups and the phenotype switch of the same species might
both lead to the phenomenon. According to literature, total cell
concentration detected by FCM was commonly divided into
two distinct clusters: high nucleic acid bacteria (HNA) and low
nucleic acid bacteria (LNA). LNA cells are considerably much
smaller than HNA cells and harder to cultivate (Lautenschlager
et al., 2010; Wang et al., 2009). Lautenschlager observed a clear
shift from more HNA bacteria to more LNA bacteria in the serial
filtration processes (Lautenschlager et al.,2014).In our study,
the coal–sand filter and GAC tank might preferably remove
the cultivable HNA with lager cell size as well and resulted in the
decrease of R2A cultivable bacteria proportion. Low temperature
in winter months might also preferably restrain the growth of
cultivable bacteria and even induce the viable-but-nonculturable
(VBNC) stages of some species. Laboratory studies with pure
cultures have shown that low nutrient and low temperature
have been shown to be the main causes of the induction of
VBNC stages in some bacteria strains, including indicator
organisms such as Escherichia coliand water-borne pathogens
such as Vibriocholerae (Mizunoe etal., 2000;Xu et al., 1982).
Disinfection was another environment pressure that causes
the adoption of a VBNC state. Hoefel ever found that
Stenotrophomonas maltophila was initially cultivable by HPC in
raw water and settled water, but undetected by HPC after
disinfection whilst remaining active (Hoefel et al., 2005).
Decreased R2A cultivability of total bacteria might result in a
general underestimation of biomass with HPC method. Further
detailed investigation targeted on discrimination and sorting of
different bacteria groups by FCM combining other molecular
methods would allow a better understanding of microbial
biomass fluctuation.
2.2. Diversity and composition of the microbial community
The bacterial 16S rRNA gene V1–V3 region of all bulk water and
biofilm DNA samples was analyzed by 454-pyrosequencing.
After quality filtration of the raw data, 62,399 effective sequences
(8405–974 per sample) were obtained in total and 1615 OTUs
were assigned. To fairly compare the 30 samples at the same
sequencing depth, 950 sequences were randomly picked
from each sample for further analysis. The averaged Good's
coverage of each sampling group ranged from 90% to 92%
indicating the majority of bacterial communities had
been well covered. The averaged alpha diversity indices of
Shannon, Chao 1 were highest in the CB samples and lowest
either in CE or FW samples (Appendix A Table S3), indicating
higher richness and bacterial diversity within GAC biofilms
than in bulk water samples.
A total of 30 phyla were detected across all of the
biofilm and water samples (Fig. 3a). Proteobacteria (53.3%),
Bacteroidetes(10.7%), Firmicutes (9.9%) and Actinobacteria
(8.4%), Planctomycetes (7.3%), and Cyanobacteria (5.0%) dominated
in the water samples, constituting about 82.3% of the total
sequences in all water samples. Proteobacteria, Bacteroidetes and
Actinobacteria are the three most common dominating phyla in
drinking water systems (Lautenschlager et al., 2014; Lin et al.,
2014; Pinto et al., 2012).
In comparison to water samples, the three dominant phyla
in carbon biofilm were Proteobacteria(48.3%),Acidobacteria
(22.9%) and Planctomycetes (16.3%). Acidobacteria was also abun-
dant but not that dominant in the biofilm samples of other
studies (Pinto et al., 2012; Liao et al., 2013). It is a newly devised
phylum with 26 recognized subdivisions. Bacteria in this
phylum are widely distributed and abundant in soils and
freshwater sediments, but numbers of cultured representatives
are very limited (Newton et al., 2011).Genomic analyses of
some isolated strains from the phylum indicated that they are
best adapted to low-nutrient conditions, capable of utilizing
diverse polysaccharides,and even involved in biofilm forma-
tion (Rawat et al., 2012;Ward et al., 2009).In our study, more
than 80% of Acidobacteria in the GAC biofilm samples belonged
to the class of Chloracidobacteria.However, still little is known
about their metabolic and genetic functions.
The Cyanobacteria population was also abundant with
averaged relative abundance of about 5.0% in water samples,
but this decreased to 0.2% in the GAC biofilm samples, which
contradicted a recent study that showed Cyanobacteria was
more abundant in biofilm sample than in water samples
(Lin et al., 2014).Whereas other observations were consis-
tent that Cyanobacteria was more abundant in bulk water
(Lautenschlager et al., 2014; Pinto et al., 2012).
The dominant subclasses of Proteobacteria were Alpha-
proteobacteria,Betaproteobacteria,Gammaproteobacteriaand
Deltaproteobacteria (Fig.3b). Along the treatment process
flow, the relative abundances of Alphaproteobacteriaand
Betaproteobacteria decreased from 41.8% and 12.4% in raw
Fig. 2 – The percentage of R2A cultivable cells (heterotrophic
plate count/total cell concentration) along treatment
processes (* indicates no cultivable bacteria (HPCs) was
detected in the sample). HPCs: heterotrophic plate count.
24 J O U R N A L O F E N V I R O N M E N T A L S C I E N C E S 5 1 ( 2 0 1 7 ) 2 1 – 3 0
Secure Best Marks with AI Grader
Need help grading? Try our AI Grader for instant feedback on your assignments.
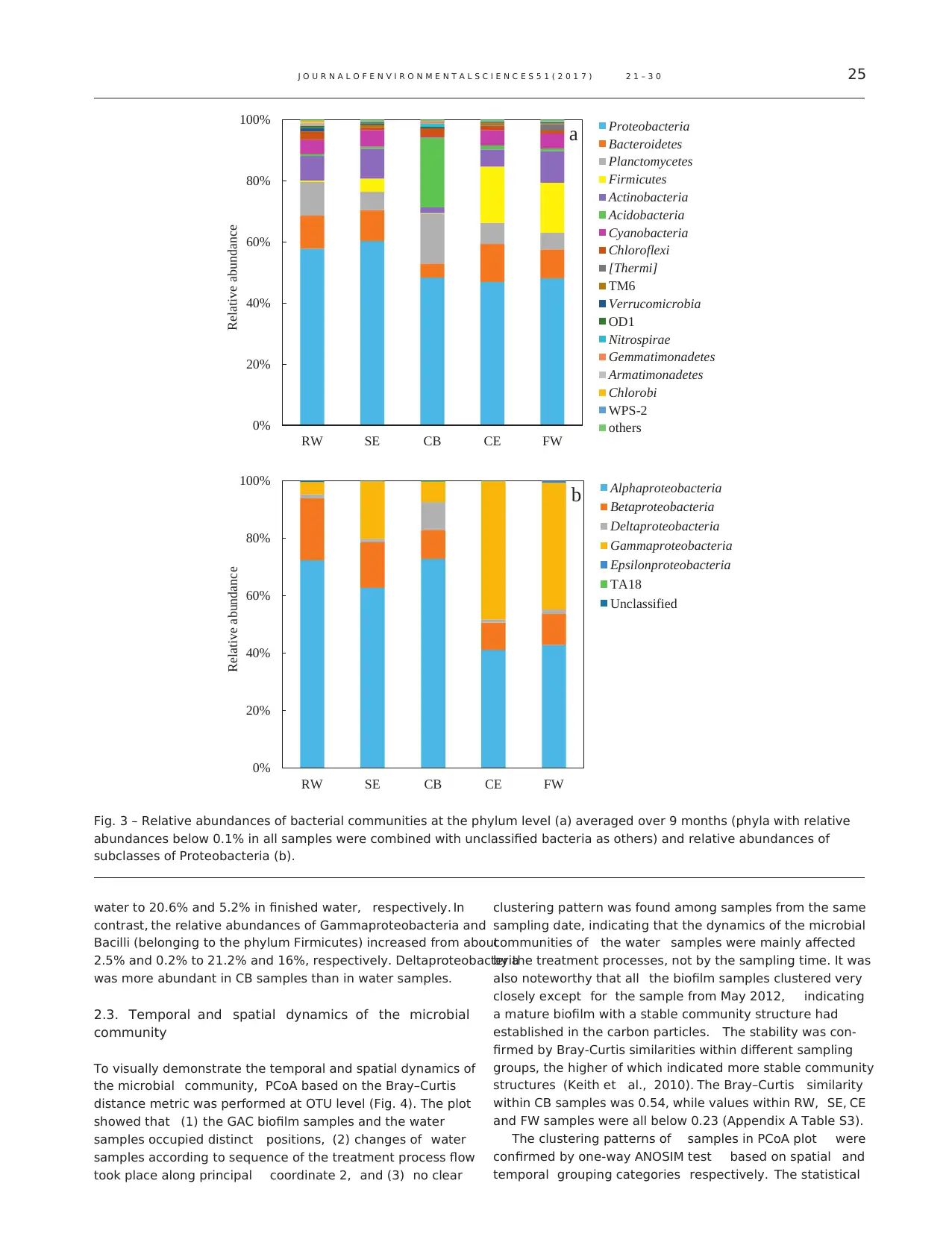
water to 20.6% and 5.2% in finished water, respectively. In
contrast, the relative abundances of Gammaproteobacteria and
Bacilli (belonging to the phylum Firmicutes) increased from about
2.5% and 0.2% to 21.2% and 16%, respectively. Deltaproteobacteria
was more abundant in CB samples than in water samples.
2.3. Temporal and spatial dynamics of the microbial
community
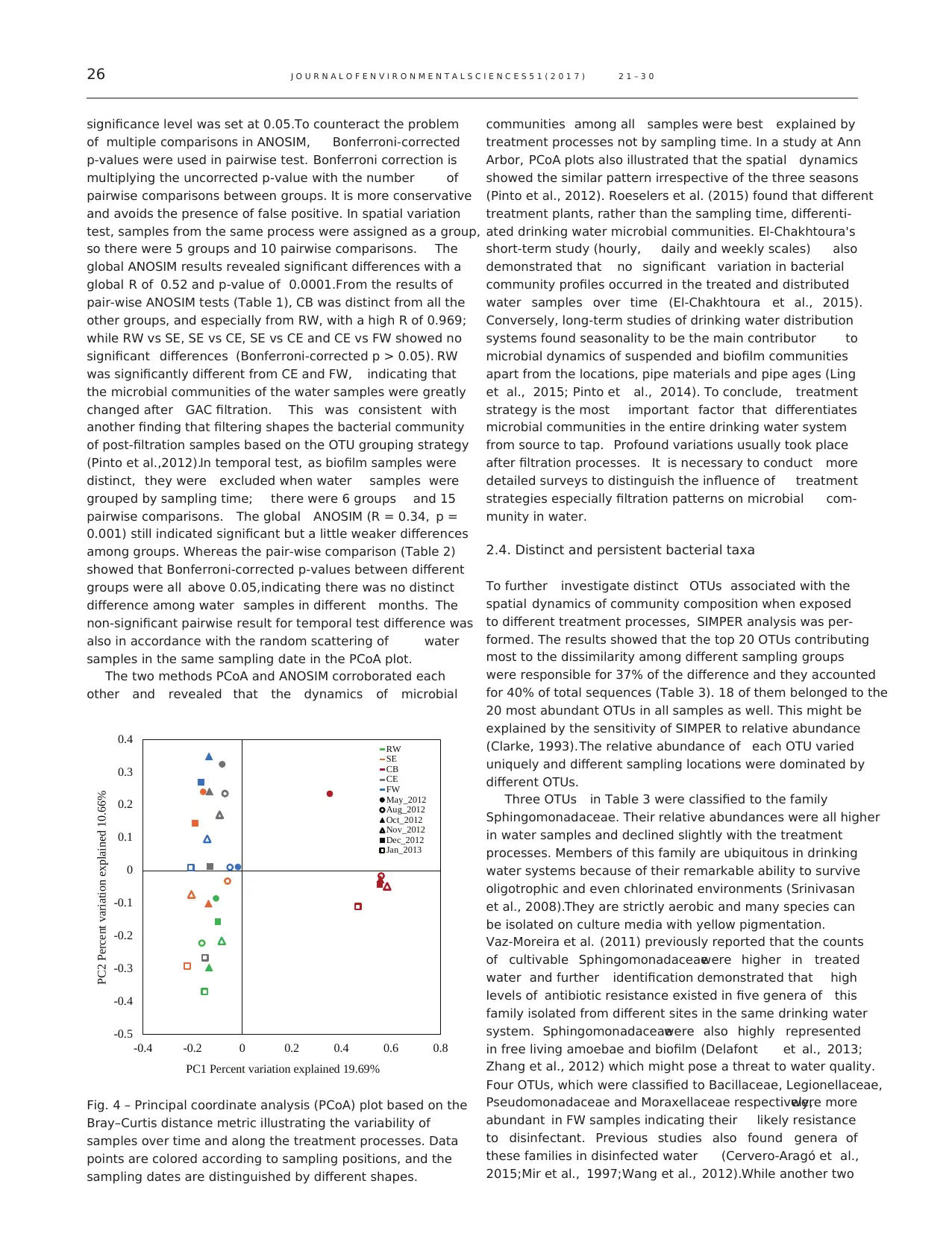
To visually demonstrate the temporal and spatial dynamics of
the microbial community, PCoA based on the Bray–Curtis
distance metric was performed at OTU level (Fig. 4). The plot
showed that (1) the GAC biofilm samples and the water
samples occupied distinct positions, (2) changes of water
samples according to sequence of the treatment process flow
took place along principal coordinate 2, and (3) no clear
clustering pattern was found among samples from the same
sampling date, indicating that the dynamics of the microbial
communities of the water samples were mainly affected
by the treatment processes, not by the sampling time. It was
also noteworthy that all the biofilm samples clustered very
closely except for the sample from May 2012, indicating
a mature biofilm with a stable community structure had
established in the carbon particles. The stability was con-
firmed by Bray-Curtis similarities within different sampling
groups, the higher of which indicated more stable community
structures (Keith et al., 2010). The Bray–Curtis similarity
within CB samples was 0.54, while values within RW, SE, CE
and FW samples were all below 0.23 (Appendix A Table S3).
The clustering patterns of samples in PCoA plot were
confirmed by one-way ANOSIM test based on spatial and
temporal grouping categories respectively. The statistical
0%
20%
40%
60%
80%
100%
RW SE CB CE FW
Relative abundance
a Proteobacteria
Bacteroidetes
Planctomycetes
Firmicutes
Actinobacteria
Acidobacteria
Cyanobacteria
Chloroflexi
[Thermi]
TM6
Verrucomicrobia
OD1
Nitrospirae
Gemmatimonadetes
Armatimonadetes
Chlorobi
WPS-2
others
0%
20%
40%
60%
80%
100%
RW SE CB CE FW
Relative abundance
b Alphaproteobacteria
Betaproteobacteria
Deltaproteobacteria
Gammaproteobacteria
Epsilonproteobacteria
TA18
Unclassified
Fig. 3 – Relative abundances of bacterial communities at the phylum level (a) averaged over 9 months (phyla with relative
abundances below 0.1% in all samples were combined with unclassified bacteria as others) and relative abundances of
subclasses of Proteobacteria (b).
25J O U R N A L O F E N V I R O N M E N T A L S C I E N C E S 5 1 ( 2 0 1 7 ) 2 1 – 3 0
contrast, the relative abundances of Gammaproteobacteria and
Bacilli (belonging to the phylum Firmicutes) increased from about
2.5% and 0.2% to 21.2% and 16%, respectively. Deltaproteobacteria
was more abundant in CB samples than in water samples.
2.3. Temporal and spatial dynamics of the microbial
community
To visually demonstrate the temporal and spatial dynamics of
the microbial community, PCoA based on the Bray–Curtis
distance metric was performed at OTU level (Fig. 4). The plot
showed that (1) the GAC biofilm samples and the water
samples occupied distinct positions, (2) changes of water
samples according to sequence of the treatment process flow
took place along principal coordinate 2, and (3) no clear
clustering pattern was found among samples from the same
sampling date, indicating that the dynamics of the microbial
communities of the water samples were mainly affected
by the treatment processes, not by the sampling time. It was
also noteworthy that all the biofilm samples clustered very
closely except for the sample from May 2012, indicating
a mature biofilm with a stable community structure had
established in the carbon particles. The stability was con-
firmed by Bray-Curtis similarities within different sampling
groups, the higher of which indicated more stable community
structures (Keith et al., 2010). The Bray–Curtis similarity
within CB samples was 0.54, while values within RW, SE, CE
and FW samples were all below 0.23 (Appendix A Table S3).
The clustering patterns of samples in PCoA plot were
confirmed by one-way ANOSIM test based on spatial and
temporal grouping categories respectively. The statistical
0%
20%
40%
60%
80%
100%
RW SE CB CE FW
Relative abundance
a Proteobacteria
Bacteroidetes
Planctomycetes
Firmicutes
Actinobacteria
Acidobacteria
Cyanobacteria
Chloroflexi
[Thermi]
TM6
Verrucomicrobia
OD1
Nitrospirae
Gemmatimonadetes
Armatimonadetes
Chlorobi
WPS-2
others
0%
20%
40%
60%
80%
100%
RW SE CB CE FW
Relative abundance
b Alphaproteobacteria
Betaproteobacteria
Deltaproteobacteria
Gammaproteobacteria
Epsilonproteobacteria
TA18
Unclassified
Fig. 3 – Relative abundances of bacterial communities at the phylum level (a) averaged over 9 months (phyla with relative
abundances below 0.1% in all samples were combined with unclassified bacteria as others) and relative abundances of
subclasses of Proteobacteria (b).
25J O U R N A L O F E N V I R O N M E N T A L S C I E N C E S 5 1 ( 2 0 1 7 ) 2 1 – 3 0
significance level was set at 0.05.To counteract the problem
of multiple comparisons in ANOSIM, Bonferroni-corrected
p-values were used in pairwise test. Bonferroni correction is
multiplying the uncorrected p-value with the number of
pairwise comparisons between groups. It is more conservative
and avoids the presence of false positive. In spatial variation
test, samples from the same process were assigned as a group,
so there were 5 groups and 10 pairwise comparisons. The
global ANOSIM results revealed significant differences with a
global R of 0.52 and p-value of 0.0001.From the results of
pair-wise ANOSIM tests (Table 1), CB was distinct from all the
other groups, and especially from RW, with a high R of 0.969;
while RW vs SE, SE vs CE, SE vs CE and CE vs FW showed no
significant differences (Bonferroni-corrected p > 0.05). RW
was significantly different from CE and FW, indicating that
the microbial communities of the water samples were greatly
changed after GAC filtration. This was consistent with
another finding that filtering shapes the bacterial community
of post-filtration samples based on the OTU grouping strategy
(Pinto et al.,2012).In temporal test, as biofilm samples were
distinct, they were excluded when water samples were
grouped by sampling time; there were 6 groups and 15
pairwise comparisons. The global ANOSIM (R = 0.34, p =
0.001) still indicated significant but a little weaker differences
among groups. Whereas the pair-wise comparison (Table 2)
showed that Bonferroni-corrected p-values between different
groups were all above 0.05,indicating there was no distinct
difference among water samples in different months. The
non-significant pairwise result for temporal test difference was
also in accordance with the random scattering of water
samples in the same sampling date in the PCoA plot.
The two methods PCoA and ANOSIM corroborated each
other and revealed that the dynamics of microbial
communities among all samples were best explained by
treatment processes not by sampling time. In a study at Ann
Arbor, PCoA plots also illustrated that the spatial dynamics
showed the similar pattern irrespective of the three seasons
(Pinto et al., 2012). Roeselers et al. (2015) found that different
treatment plants, rather than the sampling time, differenti-
ated drinking water microbial communities. El-Chakhtoura's
short-term study (hourly, daily and weekly scales) also
demonstrated that no significant variation in bacterial
community profiles occurred in the treated and distributed
water samples over time (El-Chakhtoura et al., 2015).
Conversely, long-term studies of drinking water distribution
systems found seasonality to be the main contributor to
microbial dynamics of suspended and biofilm communities
apart from the locations, pipe materials and pipe ages (Ling
et al., 2015; Pinto et al., 2014). To conclude, treatment
strategy is the most important factor that differentiates
microbial communities in the entire drinking water system
from source to tap. Profound variations usually took place
after filtration processes. It is necessary to conduct more
detailed surveys to distinguish the influence of treatment
strategies especially filtration patterns on microbial com-
munity in water.
2.4. Distinct and persistent bacterial taxa
To further investigate distinct OTUs associated with the
spatial dynamics of community composition when exposed
to different treatment processes, SIMPER analysis was per-
formed. The results showed that the top 20 OTUs contributing
most to the dissimilarity among different sampling groups
were responsible for 37% of the difference and they accounted
for 40% of total sequences (Table 3). 18 of them belonged to the
20 most abundant OTUs in all samples as well. This might be
explained by the sensitivity of SIMPER to relative abundance
(Clarke, 1993).The relative abundance of each OTU varied
uniquely and different sampling locations were dominated by
different OTUs.
Three OTUs in Table 3 were classified to the family
Sphingomonadaceae. Their relative abundances were all higher
in water samples and declined slightly with the treatment
processes. Members of this family are ubiquitous in drinking
water systems because of their remarkable ability to survive
oligotrophic and even chlorinated environments (Srinivasan
et al., 2008).They are strictly aerobic and many species can
be isolated on culture media with yellow pigmentation.
Vaz-Moreira et al. (2011) previously reported that the counts
of cultivable Sphingomonadaceaewere higher in treated
water and further identification demonstrated that high
levels of antibiotic resistance existed in five genera of this
family isolated from different sites in the same drinking water
system. Sphingomonadaceaewere also highly represented
in free living amoebae and biofilm (Delafont et al., 2013;
Zhang et al., 2012) which might pose a threat to water quality.
Four OTUs, which were classified to Bacillaceae, Legionellaceae,
Pseudomonadaceae and Moraxellaceae respectively,were more
abundant in FW samples indicating their likely resistance
to disinfectant. Previous studies also found genera of
these families in disinfected water (Cervero-Aragó et al.,
2015;Mir et al., 1997;Wang et al., 2012).While another two
-0.5
-0.4
-0.3
-0.2
-0.1
0
0.1
0.2
0.3
0.4
-0.4 -0.2 0 0.2 0.4 0.6 0.8
PC2 Percent variation explained 10.66%
PC1 Percent variation explained 19.69%
RW
SE
CB
CE
FW
May_2012
Aug_2012
Oct_2012
Nov_2012
Dec_2012
Jan_2013
Fig. 4 – Principal coordinate analysis (PCoA) plot based on the
Bray–Curtis distance metric illustrating the variability of
samples over time and along the treatment processes. Data
points are colored according to sampling positions, and the
sampling dates are distinguished by different shapes.
26 J O U R N A L O F E N V I R O N M E N T A L S C I E N C E S 5 1 ( 2 0 1 7 ) 2 1 – 3 0
of multiple comparisons in ANOSIM, Bonferroni-corrected
p-values were used in pairwise test. Bonferroni correction is
multiplying the uncorrected p-value with the number of
pairwise comparisons between groups. It is more conservative
and avoids the presence of false positive. In spatial variation
test, samples from the same process were assigned as a group,
so there were 5 groups and 10 pairwise comparisons. The
global ANOSIM results revealed significant differences with a
global R of 0.52 and p-value of 0.0001.From the results of
pair-wise ANOSIM tests (Table 1), CB was distinct from all the
other groups, and especially from RW, with a high R of 0.969;
while RW vs SE, SE vs CE, SE vs CE and CE vs FW showed no
significant differences (Bonferroni-corrected p > 0.05). RW
was significantly different from CE and FW, indicating that
the microbial communities of the water samples were greatly
changed after GAC filtration. This was consistent with
another finding that filtering shapes the bacterial community
of post-filtration samples based on the OTU grouping strategy
(Pinto et al.,2012).In temporal test, as biofilm samples were
distinct, they were excluded when water samples were
grouped by sampling time; there were 6 groups and 15
pairwise comparisons. The global ANOSIM (R = 0.34, p =
0.001) still indicated significant but a little weaker differences
among groups. Whereas the pair-wise comparison (Table 2)
showed that Bonferroni-corrected p-values between different
groups were all above 0.05,indicating there was no distinct
difference among water samples in different months. The
non-significant pairwise result for temporal test difference was
also in accordance with the random scattering of water
samples in the same sampling date in the PCoA plot.
The two methods PCoA and ANOSIM corroborated each
other and revealed that the dynamics of microbial
communities among all samples were best explained by
treatment processes not by sampling time. In a study at Ann
Arbor, PCoA plots also illustrated that the spatial dynamics
showed the similar pattern irrespective of the three seasons
(Pinto et al., 2012). Roeselers et al. (2015) found that different
treatment plants, rather than the sampling time, differenti-
ated drinking water microbial communities. El-Chakhtoura's
short-term study (hourly, daily and weekly scales) also
demonstrated that no significant variation in bacterial
community profiles occurred in the treated and distributed
water samples over time (El-Chakhtoura et al., 2015).
Conversely, long-term studies of drinking water distribution
systems found seasonality to be the main contributor to
microbial dynamics of suspended and biofilm communities
apart from the locations, pipe materials and pipe ages (Ling
et al., 2015; Pinto et al., 2014). To conclude, treatment
strategy is the most important factor that differentiates
microbial communities in the entire drinking water system
from source to tap. Profound variations usually took place
after filtration processes. It is necessary to conduct more
detailed surveys to distinguish the influence of treatment
strategies especially filtration patterns on microbial com-
munity in water.
2.4. Distinct and persistent bacterial taxa
To further investigate distinct OTUs associated with the
spatial dynamics of community composition when exposed
to different treatment processes, SIMPER analysis was per-
formed. The results showed that the top 20 OTUs contributing
most to the dissimilarity among different sampling groups
were responsible for 37% of the difference and they accounted
for 40% of total sequences (Table 3). 18 of them belonged to the
20 most abundant OTUs in all samples as well. This might be
explained by the sensitivity of SIMPER to relative abundance
(Clarke, 1993).The relative abundance of each OTU varied
uniquely and different sampling locations were dominated by
different OTUs.
Three OTUs in Table 3 were classified to the family
Sphingomonadaceae. Their relative abundances were all higher
in water samples and declined slightly with the treatment
processes. Members of this family are ubiquitous in drinking
water systems because of their remarkable ability to survive
oligotrophic and even chlorinated environments (Srinivasan
et al., 2008).They are strictly aerobic and many species can
be isolated on culture media with yellow pigmentation.
Vaz-Moreira et al. (2011) previously reported that the counts
of cultivable Sphingomonadaceaewere higher in treated
water and further identification demonstrated that high
levels of antibiotic resistance existed in five genera of this
family isolated from different sites in the same drinking water
system. Sphingomonadaceaewere also highly represented
in free living amoebae and biofilm (Delafont et al., 2013;
Zhang et al., 2012) which might pose a threat to water quality.
Four OTUs, which were classified to Bacillaceae, Legionellaceae,
Pseudomonadaceae and Moraxellaceae respectively,were more
abundant in FW samples indicating their likely resistance
to disinfectant. Previous studies also found genera of
these families in disinfected water (Cervero-Aragó et al.,
2015;Mir et al., 1997;Wang et al., 2012).While another two
-0.5
-0.4
-0.3
-0.2
-0.1
0
0.1
0.2
0.3
0.4
-0.4 -0.2 0 0.2 0.4 0.6 0.8
PC2 Percent variation explained 10.66%
PC1 Percent variation explained 19.69%
RW
SE
CB
CE
FW
May_2012
Aug_2012
Oct_2012
Nov_2012
Dec_2012
Jan_2013
Fig. 4 – Principal coordinate analysis (PCoA) plot based on the
Bray–Curtis distance metric illustrating the variability of
samples over time and along the treatment processes. Data
points are colored according to sampling positions, and the
sampling dates are distinguished by different shapes.
26 J O U R N A L O F E N V I R O N M E N T A L S C I E N C E S 5 1 ( 2 0 1 7 ) 2 1 – 3 0

OTUs also affiliated to Moraxellaceae and abundant in CE
samples did not occur in FW samples. They were classified to
the same genus Acinetobacter,but different species. The OTU
classified to family Ellin6075 was the most abundant OTU in
CB samples (accounted for 15.9% of total sequences in CB
samples and 78.5% of Acidobacteria in all samples). Though the
phylum Acidobacteria has been poorly studied and few
cultivated representatives are known, some species of the
family Ellin6075 were isolated under selective laboratory
conditions (Crowe et al., 2014;Joseph et al., 2003) indicating
the possibility of further analysis. In water samples its relative
abundance increased slightly in the CE samples by 0.4%
compared to SE samples indicating the predominant bacteria
in biofilm might seed the effluent through sloughing of
biofilms.
Although bacterial community changed significantly at
each step of treatment processes, some OTUs were com-
monly found throughout treatment processes and over
different sampling time. OTUs that were present in at least
50% of samples at each sampling location were depicted in a
Venn diagram (Fig. 5). Six of these OTUs, which constituted
16.1% of the total sequences, were shared by all sampling
groups. According to the conceptual framework of the core
community proposed by Shade and Handelsman (2012), they
were defined persistent OTUs in the entire treatment process
flow. Three of the six OTUs were also among the 20 most
abundant OTUs in all samples. Five of the six OTUs were
classified to the class Alphaproteobacteria and the other one was
classified to the genus Chryseobacterium, belonging to the class
Flavobacteriia. Alphaproteobacteria is dominant in most drinking
water systems (El-Chakhtoura et al., 2015; Williams et al., 2004)
and has been reported to be competitive in low nutrient
freshwater with availability of degrading complex organic
compounds (Newton et al., 2011). The genus Chryseobacterium
was reported to be an amoebae-resisting bacterium in a
DWTP and the species Chryseobacterium meningosepticum
which could colonize water taps was pathogenic in humans
(Hoque et al., 2001; Thomas et al., 2008). In addition, the
number of shared OTUs among CB and CE samples was 27, 7
more than that among SE and CB samples. This supported
the finding that CE water was slightly influenced by CB
samples. Persistent OTUs in systems were usually difficult to
remove completely and remained in finished water as a
potential threat. Therefore comprehensive and systematic
studies on distinct and core OTUs will make important
contributions to the understanding of microbial characters
of drinking water systems.
3. Conclusions
This study provides a comprehensive view of the dynamics
of the microbial biomass and bacterial community during
treatment processes over time. The important findings
achieved were as follows: (1) The treatment processes and
water temperature probably had remarkable influences on
the R2A cultivability of total bacteria. (2) The variation of
bacterial communities with treatment processes rather
than the temporal fluctuation was the primary dynamic
pattern. (3) The GAC filter harbored a mature biofilm with a
stable and distinct microbial community. Profound changes
in the microbial community in water occurred after GAC
filtration. (4) Distinct bacterial taxa at each process and
persistent taxa throughout the treatment process flow were
identified.
Acknowledgments
This work was supported by the China Major Science and
Technology Program for Water Pollution Control and Treat-
ment (No. 2012ZX07404-002) and the Special Fund of State Key
Joint Laboratory of Environment Simulation and Pollution
Control (No. 14K09ESPCT).
Table 1 – ANOSIM statistics for pairwise comparisons
of samples grouped by treatment process (the global R =
0.52, p = 0.0001). *
(R, pB) CB RW SE CE
CB
RW (0.969,0.019)
SE (0.935,0.025) (0.081,1)
CE (0.874,0.026) (0.652,0.024) (0.017,1)
FW (0.738,0.022) (0.490,0.024) (0.057,1) (0.119,0.953)
* pB represents Bonferroni-corrected p-value. Values in bold indicate
a Bonferroni-corrected p-value < 0.05 (uncorrected p-values < 0.005).
Table 2 – Analysis of similarity (ANOSIM) statistics for pairwise comparisons of water samples grouped by sampling
months (the global R = 0.34, p = 0.001). *
(R, pB) May Aug Oct Nov Dec
May
Aug (0.130, 1)
Oct (0.313,0.863) (0.120,1)
Nov (0.417,0.431) (0.146,1) (0.396,1)
Dec (0.260,0.431) (0.208,1) (0.427,1) (0.208,1)
Jan (0.604,0.408) (0.500,0.453) (0.635,0.515) (0.781,0.408) (0.370,0.903)
* p B represents Bonferroni-corrected p-value. All Bonferroni-corrected p-values > 0.05 (uncorrected p-values > 0.003).
27J O U R N A L O F E N V I R O N M E N T A L S C I E N C E S 5 1 ( 2 0 1 7 ) 2 1 – 3 0
samples did not occur in FW samples. They were classified to
the same genus Acinetobacter,but different species. The OTU
classified to family Ellin6075 was the most abundant OTU in
CB samples (accounted for 15.9% of total sequences in CB
samples and 78.5% of Acidobacteria in all samples). Though the
phylum Acidobacteria has been poorly studied and few
cultivated representatives are known, some species of the
family Ellin6075 were isolated under selective laboratory
conditions (Crowe et al., 2014;Joseph et al., 2003) indicating
the possibility of further analysis. In water samples its relative
abundance increased slightly in the CE samples by 0.4%
compared to SE samples indicating the predominant bacteria
in biofilm might seed the effluent through sloughing of
biofilms.
Although bacterial community changed significantly at
each step of treatment processes, some OTUs were com-
monly found throughout treatment processes and over
different sampling time. OTUs that were present in at least
50% of samples at each sampling location were depicted in a
Venn diagram (Fig. 5). Six of these OTUs, which constituted
16.1% of the total sequences, were shared by all sampling
groups. According to the conceptual framework of the core
community proposed by Shade and Handelsman (2012), they
were defined persistent OTUs in the entire treatment process
flow. Three of the six OTUs were also among the 20 most
abundant OTUs in all samples. Five of the six OTUs were
classified to the class Alphaproteobacteria and the other one was
classified to the genus Chryseobacterium, belonging to the class
Flavobacteriia. Alphaproteobacteria is dominant in most drinking
water systems (El-Chakhtoura et al., 2015; Williams et al., 2004)
and has been reported to be competitive in low nutrient
freshwater with availability of degrading complex organic
compounds (Newton et al., 2011). The genus Chryseobacterium
was reported to be an amoebae-resisting bacterium in a
DWTP and the species Chryseobacterium meningosepticum
which could colonize water taps was pathogenic in humans
(Hoque et al., 2001; Thomas et al., 2008). In addition, the
number of shared OTUs among CB and CE samples was 27, 7
more than that among SE and CB samples. This supported
the finding that CE water was slightly influenced by CB
samples. Persistent OTUs in systems were usually difficult to
remove completely and remained in finished water as a
potential threat. Therefore comprehensive and systematic
studies on distinct and core OTUs will make important
contributions to the understanding of microbial characters
of drinking water systems.
3. Conclusions
This study provides a comprehensive view of the dynamics
of the microbial biomass and bacterial community during
treatment processes over time. The important findings
achieved were as follows: (1) The treatment processes and
water temperature probably had remarkable influences on
the R2A cultivability of total bacteria. (2) The variation of
bacterial communities with treatment processes rather
than the temporal fluctuation was the primary dynamic
pattern. (3) The GAC filter harbored a mature biofilm with a
stable and distinct microbial community. Profound changes
in the microbial community in water occurred after GAC
filtration. (4) Distinct bacterial taxa at each process and
persistent taxa throughout the treatment process flow were
identified.
Acknowledgments
This work was supported by the China Major Science and
Technology Program for Water Pollution Control and Treat-
ment (No. 2012ZX07404-002) and the Special Fund of State Key
Joint Laboratory of Environment Simulation and Pollution
Control (No. 14K09ESPCT).
Table 1 – ANOSIM statistics for pairwise comparisons
of samples grouped by treatment process (the global R =
0.52, p = 0.0001). *
(R, pB) CB RW SE CE
CB
RW (0.969,0.019)
SE (0.935,0.025) (0.081,1)
CE (0.874,0.026) (0.652,0.024) (0.017,1)
FW (0.738,0.022) (0.490,0.024) (0.057,1) (0.119,0.953)
* pB represents Bonferroni-corrected p-value. Values in bold indicate
a Bonferroni-corrected p-value < 0.05 (uncorrected p-values < 0.005).
Table 2 – Analysis of similarity (ANOSIM) statistics for pairwise comparisons of water samples grouped by sampling
months (the global R = 0.34, p = 0.001). *
(R, pB) May Aug Oct Nov Dec
May
Aug (0.130, 1)
Oct (0.313,0.863) (0.120,1)
Nov (0.417,0.431) (0.146,1) (0.396,1)
Dec (0.260,0.431) (0.208,1) (0.427,1) (0.208,1)
Jan (0.604,0.408) (0.500,0.453) (0.635,0.515) (0.781,0.408) (0.370,0.903)
* p B represents Bonferroni-corrected p-value. All Bonferroni-corrected p-values > 0.05 (uncorrected p-values > 0.003).
27J O U R N A L O F E N V I R O N M E N T A L S C I E N C E S 5 1 ( 2 0 1 7 ) 2 1 – 3 0
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

Appendix A. Supplementary data
Supplementary data to this article can be found online at
http://dx.doi.org/10.1016/j.jes.2016.05.042.
R E F E R E N C E S
Allen, M.J., Edberg, S.C., Reasoner, D.J., 2004. Heterotrophic plate
count bacteria-what is their significance in drinking water?
Int. J. Food Microbiol. 92 (3), 265–274.
Au, K., 2004. Water Treatment and Pathogen Control: Process
Efficiency in Achieving Safe Drinking-Water. IWA Publishing.
Bartram, J., Cotruvo, J., Exner, M., Fricker, C., Glasmacher, A., 2003.
Heterotrophic Plate Counts and Drinking-Water Safety: The
Significance of HPCs for Water Quality and Human Health.
IWA Publishing.
Berry, D., Xi, C., Raskin, L., 2006. Microbial ecology of drinking
water distribution systems. Curr. Opin. Biotechnol. 17 (3),
297–302.
Boe-Hansen, R., Albrechtsen, H., Arvin, E., Jørgensen, C., 2002. Bulk
water phase and biofilm growth in drinking water at low
nutrient conditions. Water Res. 36 (18), 4477–4486.
Caporaso, J.G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman,
F.D., Costello, E.K., Fierer, N., Pena, A.G., Goodrich, J.K., Gordon,
J.I., 2010. QIIME allows analysis of high-throughput
community sequencing data. Nat. Methods 7 (5), 335–336.
Carter, J.T., Rice, E.W., Buchberger, S.G., Lee, Y., 2000.
Relationships between levels of heterotrophic bacteria and
water quality parameters in a drinking water distribution
system. Water Res. 34 (5), 1495–1502.
Cervero-Aragó, S., Rodríguez-Martínez, S., Puertas-Bennasar, A.,
Araujo, R.M., 2015. Effect of common drinking water
disinfectants, chlorine and heat, on free Legionella and
amoebae-associated Legionella. PLoS One 10 (8), e134726.
Chen, C., Zhang, X., He, W., Lu, W., Han, H., 2007. Comparison of
seven kinds of drinking water treatment processes to enhance
organic material removal: a pilot test. Sci. Total Environ. 382
(1), 93–102.
Clarke, K.R., 1993. Non-parametric multivariate analyses of
changes in community structure. Aust. J. Ecol. 18, 117.
Crowe, M.A., Power, J.F., Morgan, X.C., Dunfield, P.F., Lagutin, K.,
Rijpstra, W., Vyssotski, M., Damste, J.S., Houghton, K.M., Ryan,
Table 3 – SIMPER analysis results displaying top 20 OTUs responsible for dissimilarity among different sampling groups.
No. Contrib.
%
Cumulative
%
Mean
abund. CB
Mean
abund. RW
Mean
abund. SE
Mean
abund. CE
Mean
abund. FW
Family
OTU1909 5.294 5.294 1.34% 17.05% 8.84% 4.92% 4.11% Sphingomonadaceae
OTU1597 3.693 8.987 15.89% 0.00% 0.02% 0.37% 0.04% Ellin6075
OTU1992 3.541 12.53 3.18% 0.37% 3.07% 9.86% 5.42% Weeksellaceae
OTU1441 3.147 15.67 0.00% 0.00% 1.42% 12.21% 0.46% unclassified_Bacillales
OTU2387 2.507 18.18 0.00% 0.04% 0.04% 0.02% 10.84% Bacillaceae
OTU1673 1.973 20.16 0.00% 1.28% 1.84% 1.63% 4.92% Legionellaceae
OTU1599 1.872 22.03 4.13% 4.58% 2.32% 2.32% 0.54% unclassified_Sphingomonadales
OTU1611 1.572 23.6 0.00% 0.00% 6.53% 0.28% 0.09% Sphingomonadaceae
OTU689 1.522 25.12 0.04% 1.98% 1.97% 3.16% 1.45% Gemmataceae
OTU903 1.499 26.62 0.00% 1.08% 3.11% 3.26% 0.04% Synechococcaceae
OTU1592 1.238 27.86 1.34% 0.04% 1.28% 2.40% 2.45% unclassified_HOC36
OTU1949 1.202 29.06 0.05% 0.02% 1.26% 0.26% 4.11% Pseudomonadaceae
OTU1825 1.174 30.23 0.02% 0.00% 0.12% 4.98% 0.00% Moraxellaceae
OTU1164 1.15 31.38 0.00% 3.24% 1.89% 0.47% 0.54% Pelagibacteraceae
OTU2319 1.106 32.49 0.02% 0.00% 0.47% 4.44% 0.09% Moraxellaceae
OTU1459 1.065 33.56 0.00% 1.65% 2.56% 0.70% 0.70% C111
OTU223 0.9754 34.53 0.11% 2.40% 2.37% 0.42% 0.12% Sphingomonadaceae
OTU1274 0.8756 35.41 0.07% 0.00% 0.65% 0.26% 3.18% Moraxellaceae
OTU2280 0.8225 36.23 0.04% 2.47% 1.02% 0.19% 0.05% Comamonadaceae
OTU89 0.8007 37.03 0.02% 0.02% 0.00% 2.93% 0.61% Bacillaceae
RW: raw water; SE: coal–sand filter effluent; CB: GAC particles; CE: GAC tank effluent; OTU: operational taxonomic units; SIMPER: similarity
percentage.
Fig. 5 – Venn diagram showing the numbers of shared
operational taxonomic units (OTUs) among samples from
different sampling locations (the numbers in overlaps of the
diagram indicated shared OTUs that were detected in at least
50% of samples at each of the two or more corresponding
sampling locations).
28 J O U R N A L O F E N V I R O N M E N T A L S C I E N C E S 5 1 ( 2 0 1 7 ) 2 1 – 3 0
Supplementary data to this article can be found online at
http://dx.doi.org/10.1016/j.jes.2016.05.042.
R E F E R E N C E S
Allen, M.J., Edberg, S.C., Reasoner, D.J., 2004. Heterotrophic plate
count bacteria-what is their significance in drinking water?
Int. J. Food Microbiol. 92 (3), 265–274.
Au, K., 2004. Water Treatment and Pathogen Control: Process
Efficiency in Achieving Safe Drinking-Water. IWA Publishing.
Bartram, J., Cotruvo, J., Exner, M., Fricker, C., Glasmacher, A., 2003.
Heterotrophic Plate Counts and Drinking-Water Safety: The
Significance of HPCs for Water Quality and Human Health.
IWA Publishing.
Berry, D., Xi, C., Raskin, L., 2006. Microbial ecology of drinking
water distribution systems. Curr. Opin. Biotechnol. 17 (3),
297–302.
Boe-Hansen, R., Albrechtsen, H., Arvin, E., Jørgensen, C., 2002. Bulk
water phase and biofilm growth in drinking water at low
nutrient conditions. Water Res. 36 (18), 4477–4486.
Caporaso, J.G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman,
F.D., Costello, E.K., Fierer, N., Pena, A.G., Goodrich, J.K., Gordon,
J.I., 2010. QIIME allows analysis of high-throughput
community sequencing data. Nat. Methods 7 (5), 335–336.
Carter, J.T., Rice, E.W., Buchberger, S.G., Lee, Y., 2000.
Relationships between levels of heterotrophic bacteria and
water quality parameters in a drinking water distribution
system. Water Res. 34 (5), 1495–1502.
Cervero-Aragó, S., Rodríguez-Martínez, S., Puertas-Bennasar, A.,
Araujo, R.M., 2015. Effect of common drinking water
disinfectants, chlorine and heat, on free Legionella and
amoebae-associated Legionella. PLoS One 10 (8), e134726.
Chen, C., Zhang, X., He, W., Lu, W., Han, H., 2007. Comparison of
seven kinds of drinking water treatment processes to enhance
organic material removal: a pilot test. Sci. Total Environ. 382
(1), 93–102.
Clarke, K.R., 1993. Non-parametric multivariate analyses of
changes in community structure. Aust. J. Ecol. 18, 117.
Crowe, M.A., Power, J.F., Morgan, X.C., Dunfield, P.F., Lagutin, K.,
Rijpstra, W., Vyssotski, M., Damste, J.S., Houghton, K.M., Ryan,
Table 3 – SIMPER analysis results displaying top 20 OTUs responsible for dissimilarity among different sampling groups.
No. Contrib.
%
Cumulative
%
Mean
abund. CB
Mean
abund. RW
Mean
abund. SE
Mean
abund. CE
Mean
abund. FW
Family
OTU1909 5.294 5.294 1.34% 17.05% 8.84% 4.92% 4.11% Sphingomonadaceae
OTU1597 3.693 8.987 15.89% 0.00% 0.02% 0.37% 0.04% Ellin6075
OTU1992 3.541 12.53 3.18% 0.37% 3.07% 9.86% 5.42% Weeksellaceae
OTU1441 3.147 15.67 0.00% 0.00% 1.42% 12.21% 0.46% unclassified_Bacillales
OTU2387 2.507 18.18 0.00% 0.04% 0.04% 0.02% 10.84% Bacillaceae
OTU1673 1.973 20.16 0.00% 1.28% 1.84% 1.63% 4.92% Legionellaceae
OTU1599 1.872 22.03 4.13% 4.58% 2.32% 2.32% 0.54% unclassified_Sphingomonadales
OTU1611 1.572 23.6 0.00% 0.00% 6.53% 0.28% 0.09% Sphingomonadaceae
OTU689 1.522 25.12 0.04% 1.98% 1.97% 3.16% 1.45% Gemmataceae
OTU903 1.499 26.62 0.00% 1.08% 3.11% 3.26% 0.04% Synechococcaceae
OTU1592 1.238 27.86 1.34% 0.04% 1.28% 2.40% 2.45% unclassified_HOC36
OTU1949 1.202 29.06 0.05% 0.02% 1.26% 0.26% 4.11% Pseudomonadaceae
OTU1825 1.174 30.23 0.02% 0.00% 0.12% 4.98% 0.00% Moraxellaceae
OTU1164 1.15 31.38 0.00% 3.24% 1.89% 0.47% 0.54% Pelagibacteraceae
OTU2319 1.106 32.49 0.02% 0.00% 0.47% 4.44% 0.09% Moraxellaceae
OTU1459 1.065 33.56 0.00% 1.65% 2.56% 0.70% 0.70% C111
OTU223 0.9754 34.53 0.11% 2.40% 2.37% 0.42% 0.12% Sphingomonadaceae
OTU1274 0.8756 35.41 0.07% 0.00% 0.65% 0.26% 3.18% Moraxellaceae
OTU2280 0.8225 36.23 0.04% 2.47% 1.02% 0.19% 0.05% Comamonadaceae
OTU89 0.8007 37.03 0.02% 0.02% 0.00% 2.93% 0.61% Bacillaceae
RW: raw water; SE: coal–sand filter effluent; CB: GAC particles; CE: GAC tank effluent; OTU: operational taxonomic units; SIMPER: similarity
percentage.
Fig. 5 – Venn diagram showing the numbers of shared
operational taxonomic units (OTUs) among samples from
different sampling locations (the numbers in overlaps of the
diagram indicated shared OTUs that were detected in at least
50% of samples at each of the two or more corresponding
sampling locations).
28 J O U R N A L O F E N V I R O N M E N T A L S C I E N C E S 5 1 ( 2 0 1 7 ) 2 1 – 3 0

J., 2014. Pyrinomonas methylaliphatogenes gen. nov., sp. nov., a
novel group 4 thermophilic member of the phylum
Acidobacteria from geothermal soils. Int. J. Syst. Evol. Microbiol.
64 (1), 220–227.
Delafont, V., Brouke, A., Bouchon, D., Moulin, L., Héchard, Y., 2013.
Microbiome of free-living amoebae isolated from drinking
water. Water Res. 47 (19), 6958–6965.
Eichler, S., Christen, R., Höltje, C., Westphal, P., Bötel, J., Brettar,
I., Mehling, A., Höfle, M.G., 2006. Composition and dynamics
of bacterial communities of a drinking water supply system
as assessed by RNA-and DNA-based 16S rRNA gene
fingerprinting. Appl. Environ. Microbiol. 72 (3), 1858–1872.
El-Chakhtoura, J., Prest, E., Saikaly, P., van Loosdrecht, M.,
Hammes, F., Vrouwenvelder, H., 2015. Dynamics of bacterial
communities before and after distribution in a full-scale
drinking water network. Water Res. 180–190.
Hammes, F., Berney, M., Wang, Y., Vital, M., Köster, O., Egli, T.,
2008. Flow-cytometric total bacterial cell counts as a
descriptive microbiological parameter for drinking water
treatment processes. Water Res. 42 (1), 269–277.
Henne, K., Kahlisch, L., Höfle, M.G., Brettar, I., 2013. Seasonal
dynamics of bacterial community structure and composition
in cold and hot drinking water derived from surface water
reservoirs. Water Res. 47 (15), 5614–5630.
Hoefel, D., Monis, P.T., Grooby, W.L., Andrews, S., Saint, C.P., 2005.
Profiling bacterial survival through a water treatment process
and subsequent distribution system. J. Appl. Microbiol. 99 (1),
175–186.
Holinger, E.P., Ross, K.A., Robertson, C.E., Stevens, M.J., Harris, J.K.,
Pace, N.R., 2014. Molecular analysis of point-of-use municipal
drinking water microbiology. Water Res. 49, 225–235.
Hoque, S.N., Graham, J., Kaufmann, M.E., Tabaqchali, S., 2001.
Chryseobacterium (Flavobacterium) meningosepticum
outbreak associated with colonization of water taps in a
neonatal intensive care unit. J. Hosp. Infect. 47 (3), 188–192.
Hwang, C., Ling, F., Andersen, G.L., LeChevallier, M.W., Liu, W.,
2012. Microbial community dynamics of an urban drinking
water distribution system subjected to phases of
chloramination and chlorination treatments. Appl. Environ.
Microbiol. 78 (22), 7856–7865.
Isaac-Renton, J., Moorehead, W., Ross, A., 1996. Longitudinal
studies of Giardia contamination in two community drinking
water supplies: cyst levels, parasite viability, and health
impact. Appl. Environ. Microbiol. 62 (1), 47–54.
Joseph, S.J., Hugenholtz, P., Sangwan, P., Osborne, C.A., Janssen,
P.H., 2003. Laboratory cultivation of widespread and previously
uncultured soil bacteria. Appl. Environ. Microbiol. 69 (12),
7210–7215.
Keith, A.R., Bailey, J.K., Whitham, T.G., 2010. A genetic basis to
community repeatability and stability. Ecology 91 (11), 3398–3406.
Kwon, S., Moon, E., Kim, T., Hong, S., Park, H., 2011. Pyrosequencing
demonstrated complex microbial communities in a membrane
filtration system for a drinking water treatment plant. Microbes
Environ. 26 (2), 149–155.
Lautenschlager, K., Boon, N., Wang, Y., Egli, T., Hammes, F., 2010.
Overnight stagnation of drinking water in household taps
induces microbial growth and changes in community
composition. Water Res. 44 (17), 4868–4877.
Lautenschlager, K., Hwang, C., Ling, F., Liu, W., Boon, N., Köster, O.,
Egli, T., Hammes, F., 2014. Abundance and composition
of indigenous bacterial communities in a multi-step
biofiltration-based drinking water treatment plant. Water Res.
62, 40–52.
LeChevallier, M.W., Schulz, W., Lee, R.G., 1991. Bacterial nutrients in
drinking water. Appl. Environ. Microbiol. 57 (3), 857–862.
Liao, X., Chen, C., Wang, Z., Wan, R., Chang, C., Zhang, X., Xie, S.,
2013. Changes of biomass and bacterial communities in
biological activated carbon filters for drinking water treatment.
Process Biochem. 48 (2), 312–316.
Lin, W., Yu, Z., Zhang, H., Thompson, I.P., 2014. Diversity and
dynamics of microbial communities at each step of treatment
plant for potable water generation. Water Res. 52, 218–230.
Ling, F., Hwang, C., LeChevallier, M.W., Andersen, G.L., Liu, W.,
2016. Core-satellite populations and seasonality of water
meter biofilms in a metropolitan drinking water distribution
system. ISME J. 10 (3), 582–595.
Liu, W., Wu, H., Wang, Z., Ong, S.L., Hu, J.Y., Ng, W.J., 2002.
Investigation of assimilable organic carbon (AOC) and bacterial
regrowth in drinking water distribution system. Water Res. 36
(4), 891–898.
Lu, P., Zhang, X., Zhang, C., Niu, Z., Xie, S., Chen, C., 2014.
Biostability in distribution systems in one city in southern
China: characteristics, modeling and control strategy.
J. Environ. Sci. 26 (2), 323–331.
Mi, Z., Dai, Y., Xie, S., Chen, C., Zhang, X., 2015. Impact of
disinfection on drinking water biofilm bacterial community.
37 (11), 200–205.
Mir, J., Morato, J., Ribas, F., 1997. Resistance to chlorine of
freshwater bacterial strains. J. Appl. Microbiol. 82 (1), 7–18.
Mizunoe, Y., Wai, S.N., Ishikawa, T., Takade, A., Yoshida, S., 2000.
Resuscitation of viable but nonculturable cells of Vibrio
parahaemolyticus induced at low temperature under starvation.
FEMS Microbiol. Lett. 186 (1), 115–120.
Newton, R.J., Jones, S.E., Eiler, A., McMahon, K.D., Bertilsson, S.,
2011. A guide to the natural history of freshwater lake bacteria.
Microbiol. Mol. Biol. Rev. 1 (75), 14–49.
Pinto, A.J., Xi, C., Raskin, L., 2012. Bacterial community structure in
the drinking water microbiome is governed by filtration
processes. Environ. Sci. Technol. 46 (16), 8851–8859.
Pinto, A.J., Schroeder, J., Lunn, M., Sloan, W., Raskin, L., 2014.
Spatial–temporal survey and occupancy-abundance modeling
to predict bacterial community dynamics in the drinking water
microbiome. Mbio 5 (3), 1–13.
Rawat, S.R., Männistö, M.K., Bromberg, Y., Häggblom, M.M., 2012.
Comparative genomic and physiological analysis provides
insights into the role of Acidobacteria in organic carbon
utilization in Arctic tundra soils. FEMS Microbiol. Ecol. 82 (2),
341–355.
Roeselers, G., Coolen, J., Wielen, P.W., Jaspers, M.C., Atsma, A.,
Graaf, B., Schuren, F., 2015. Microbial biogeography of drinking
water: patterns in phylogenetic diversity across space and
time. Environ. Microbiol. 17 (7), 2505–2514.
Shade, A., Handelsman, J., 2012. Beyond the Venn diagram: the
hunt for a core microbiome. Environ. Microbiol. 14 (1), 4–12.
Sharp, E.L., Parsons, S.A., Jefferson, B., 2006. Seasonal variations in
natural organic matter and its impact on coagulation in water
treatment. Sci. Total Environ. 363 (1), 183–194.
Srinivasan, S., Harrington, G.W., Xagoraraki, I., Goel, R., 2008.
Factors affecting bulk to total bacteria ratio in drinking water
distribution systems. Water Res. 42 (13), 3393–3404.
Thomas, V., Loret, J.F., Jousset, M., Greub, G., 2008. Biodiversity
of amoebae and amoebae-resisting bacteria in a drinking
water treatment plant. Environ. Microbiol. 10 (10),
2728–2745.
Vaz-Moreira, I., Nunes, O.C., Manaia, C.M., 2011. Diversity and
antibiotic resistance patterns of Sphingomonadaceae isolates
from drinking water. Appl. Environ. Microbiol. 77 (16),
5697–5706.
Wang, Y., Hammes, F., Boon, N., Chami, M., Egli, T., 2009. Isolation
and characterization of low nucleic acid (LNA)-content
bacteria. ISME J. 3 (8), 889–902.
Wang, H., Edwards, M., Falkinham, J.O., Pruden, A., 2012.
Molecular survey of the occurrence of Legionella spp.,
Mycobacterium spp., Pseudomonas aeruginosa, and amoeba hosts
in two chloraminated drinking water distribution systems.
Appl. Environ. Microbiol. 78 (17), 6285–6294.
Wang, H., Pryor, M.A., Edwards, M.A., Falkinham, J.O., Pruden, A.,
2013. Effect of GAC pre-treatment and disinfectant on
29J O U R N A L O F E N V I R O N M E N T A L S C I E N C E S 5 1 ( 2 0 1 7 ) 2 1 – 3 0
novel group 4 thermophilic member of the phylum
Acidobacteria from geothermal soils. Int. J. Syst. Evol. Microbiol.
64 (1), 220–227.
Delafont, V., Brouke, A., Bouchon, D., Moulin, L., Héchard, Y., 2013.
Microbiome of free-living amoebae isolated from drinking
water. Water Res. 47 (19), 6958–6965.
Eichler, S., Christen, R., Höltje, C., Westphal, P., Bötel, J., Brettar,
I., Mehling, A., Höfle, M.G., 2006. Composition and dynamics
of bacterial communities of a drinking water supply system
as assessed by RNA-and DNA-based 16S rRNA gene
fingerprinting. Appl. Environ. Microbiol. 72 (3), 1858–1872.
El-Chakhtoura, J., Prest, E., Saikaly, P., van Loosdrecht, M.,
Hammes, F., Vrouwenvelder, H., 2015. Dynamics of bacterial
communities before and after distribution in a full-scale
drinking water network. Water Res. 180–190.
Hammes, F., Berney, M., Wang, Y., Vital, M., Köster, O., Egli, T.,
2008. Flow-cytometric total bacterial cell counts as a
descriptive microbiological parameter for drinking water
treatment processes. Water Res. 42 (1), 269–277.
Henne, K., Kahlisch, L., Höfle, M.G., Brettar, I., 2013. Seasonal
dynamics of bacterial community structure and composition
in cold and hot drinking water derived from surface water
reservoirs. Water Res. 47 (15), 5614–5630.
Hoefel, D., Monis, P.T., Grooby, W.L., Andrews, S., Saint, C.P., 2005.
Profiling bacterial survival through a water treatment process
and subsequent distribution system. J. Appl. Microbiol. 99 (1),
175–186.
Holinger, E.P., Ross, K.A., Robertson, C.E., Stevens, M.J., Harris, J.K.,
Pace, N.R., 2014. Molecular analysis of point-of-use municipal
drinking water microbiology. Water Res. 49, 225–235.
Hoque, S.N., Graham, J., Kaufmann, M.E., Tabaqchali, S., 2001.
Chryseobacterium (Flavobacterium) meningosepticum
outbreak associated with colonization of water taps in a
neonatal intensive care unit. J. Hosp. Infect. 47 (3), 188–192.
Hwang, C., Ling, F., Andersen, G.L., LeChevallier, M.W., Liu, W.,
2012. Microbial community dynamics of an urban drinking
water distribution system subjected to phases of
chloramination and chlorination treatments. Appl. Environ.
Microbiol. 78 (22), 7856–7865.
Isaac-Renton, J., Moorehead, W., Ross, A., 1996. Longitudinal
studies of Giardia contamination in two community drinking
water supplies: cyst levels, parasite viability, and health
impact. Appl. Environ. Microbiol. 62 (1), 47–54.
Joseph, S.J., Hugenholtz, P., Sangwan, P., Osborne, C.A., Janssen,
P.H., 2003. Laboratory cultivation of widespread and previously
uncultured soil bacteria. Appl. Environ. Microbiol. 69 (12),
7210–7215.
Keith, A.R., Bailey, J.K., Whitham, T.G., 2010. A genetic basis to
community repeatability and stability. Ecology 91 (11), 3398–3406.
Kwon, S., Moon, E., Kim, T., Hong, S., Park, H., 2011. Pyrosequencing
demonstrated complex microbial communities in a membrane
filtration system for a drinking water treatment plant. Microbes
Environ. 26 (2), 149–155.
Lautenschlager, K., Boon, N., Wang, Y., Egli, T., Hammes, F., 2010.
Overnight stagnation of drinking water in household taps
induces microbial growth and changes in community
composition. Water Res. 44 (17), 4868–4877.
Lautenschlager, K., Hwang, C., Ling, F., Liu, W., Boon, N., Köster, O.,
Egli, T., Hammes, F., 2014. Abundance and composition
of indigenous bacterial communities in a multi-step
biofiltration-based drinking water treatment plant. Water Res.
62, 40–52.
LeChevallier, M.W., Schulz, W., Lee, R.G., 1991. Bacterial nutrients in
drinking water. Appl. Environ. Microbiol. 57 (3), 857–862.
Liao, X., Chen, C., Wang, Z., Wan, R., Chang, C., Zhang, X., Xie, S.,
2013. Changes of biomass and bacterial communities in
biological activated carbon filters for drinking water treatment.
Process Biochem. 48 (2), 312–316.
Lin, W., Yu, Z., Zhang, H., Thompson, I.P., 2014. Diversity and
dynamics of microbial communities at each step of treatment
plant for potable water generation. Water Res. 52, 218–230.
Ling, F., Hwang, C., LeChevallier, M.W., Andersen, G.L., Liu, W.,
2016. Core-satellite populations and seasonality of water
meter biofilms in a metropolitan drinking water distribution
system. ISME J. 10 (3), 582–595.
Liu, W., Wu, H., Wang, Z., Ong, S.L., Hu, J.Y., Ng, W.J., 2002.
Investigation of assimilable organic carbon (AOC) and bacterial
regrowth in drinking water distribution system. Water Res. 36
(4), 891–898.
Lu, P., Zhang, X., Zhang, C., Niu, Z., Xie, S., Chen, C., 2014.
Biostability in distribution systems in one city in southern
China: characteristics, modeling and control strategy.
J. Environ. Sci. 26 (2), 323–331.
Mi, Z., Dai, Y., Xie, S., Chen, C., Zhang, X., 2015. Impact of
disinfection on drinking water biofilm bacterial community.
37 (11), 200–205.
Mir, J., Morato, J., Ribas, F., 1997. Resistance to chlorine of
freshwater bacterial strains. J. Appl. Microbiol. 82 (1), 7–18.
Mizunoe, Y., Wai, S.N., Ishikawa, T., Takade, A., Yoshida, S., 2000.
Resuscitation of viable but nonculturable cells of Vibrio
parahaemolyticus induced at low temperature under starvation.
FEMS Microbiol. Lett. 186 (1), 115–120.
Newton, R.J., Jones, S.E., Eiler, A., McMahon, K.D., Bertilsson, S.,
2011. A guide to the natural history of freshwater lake bacteria.
Microbiol. Mol. Biol. Rev. 1 (75), 14–49.
Pinto, A.J., Xi, C., Raskin, L., 2012. Bacterial community structure in
the drinking water microbiome is governed by filtration
processes. Environ. Sci. Technol. 46 (16), 8851–8859.
Pinto, A.J., Schroeder, J., Lunn, M., Sloan, W., Raskin, L., 2014.
Spatial–temporal survey and occupancy-abundance modeling
to predict bacterial community dynamics in the drinking water
microbiome. Mbio 5 (3), 1–13.
Rawat, S.R., Männistö, M.K., Bromberg, Y., Häggblom, M.M., 2012.
Comparative genomic and physiological analysis provides
insights into the role of Acidobacteria in organic carbon
utilization in Arctic tundra soils. FEMS Microbiol. Ecol. 82 (2),
341–355.
Roeselers, G., Coolen, J., Wielen, P.W., Jaspers, M.C., Atsma, A.,
Graaf, B., Schuren, F., 2015. Microbial biogeography of drinking
water: patterns in phylogenetic diversity across space and
time. Environ. Microbiol. 17 (7), 2505–2514.
Shade, A., Handelsman, J., 2012. Beyond the Venn diagram: the
hunt for a core microbiome. Environ. Microbiol. 14 (1), 4–12.
Sharp, E.L., Parsons, S.A., Jefferson, B., 2006. Seasonal variations in
natural organic matter and its impact on coagulation in water
treatment. Sci. Total Environ. 363 (1), 183–194.
Srinivasan, S., Harrington, G.W., Xagoraraki, I., Goel, R., 2008.
Factors affecting bulk to total bacteria ratio in drinking water
distribution systems. Water Res. 42 (13), 3393–3404.
Thomas, V., Loret, J.F., Jousset, M., Greub, G., 2008. Biodiversity
of amoebae and amoebae-resisting bacteria in a drinking
water treatment plant. Environ. Microbiol. 10 (10),
2728–2745.
Vaz-Moreira, I., Nunes, O.C., Manaia, C.M., 2011. Diversity and
antibiotic resistance patterns of Sphingomonadaceae isolates
from drinking water. Appl. Environ. Microbiol. 77 (16),
5697–5706.
Wang, Y., Hammes, F., Boon, N., Chami, M., Egli, T., 2009. Isolation
and characterization of low nucleic acid (LNA)-content
bacteria. ISME J. 3 (8), 889–902.
Wang, H., Edwards, M., Falkinham, J.O., Pruden, A., 2012.
Molecular survey of the occurrence of Legionella spp.,
Mycobacterium spp., Pseudomonas aeruginosa, and amoeba hosts
in two chloraminated drinking water distribution systems.
Appl. Environ. Microbiol. 78 (17), 6285–6294.
Wang, H., Pryor, M.A., Edwards, M.A., Falkinham, J.O., Pruden, A.,
2013. Effect of GAC pre-treatment and disinfectant on
29J O U R N A L O F E N V I R O N M E N T A L S C I E N C E S 5 1 ( 2 0 1 7 ) 2 1 – 3 0

microbial community structure and opportunistic pathogen
occurrence. Water Res. 47 (15), 5760–5772.
Ward, N.L., Challacombe, J.F., Janssen, P.H., Henrissat, B.,
Coutinho, P.M., Wu, M., Xie, G., Haft, D.H., Sait, M., Badger, J.,
2009. Three genomes from the phylum Acidobacteria provide
insight into the lifestyles of these microorganisms in soils.
Appl. Environ. Microbiol. 75 (7), 2046–2056.
Williams, M.M., Domingo, J., Meckes, M.C., Kelty, C.A., Rochon,
H.S., 2004. Phylogenetic diversity of drinking water bacteria in
a distribution system simulator. J. Appl. Microbiol. 96 (5),
954–964.
Xu, H., Roberts, N., Singleton, F.L., Attwell, R.W., Grimes, D.J.,
Colwell, R.R., 1982. Survival and viability of nonculturable
Escherichia coli and Vibrio cholerae in the estuarine and marine
environment. Microb. Ecol. 8 (4), 313–323.
Zhang, M., Liu, W., Nie, X., Li, C., Gu, J., Zhang, C., 2012. Molecular
analysis of bacterial communities in biofilms of a drinking
water clearwell. Microbes Environ. 27 (4), 443.
Zhang, D., Li, W., Gong, H., Zhang, L., Gong, X., Liu, B., 2013.
Evaluation of long term stability of seeded bacteria in a
bio-enhanced activated carbon filter used for treating
drinking water. Int. Biodeterior. Biodegrad. 85, 701–708 .
30 J O U R N A L O F E N V I R O N M E N T A L S C I E N C E S 5 1 ( 2 0 1 7 ) 2 1 – 3 0
occurrence. Water Res. 47 (15), 5760–5772.
Ward, N.L., Challacombe, J.F., Janssen, P.H., Henrissat, B.,
Coutinho, P.M., Wu, M., Xie, G., Haft, D.H., Sait, M., Badger, J.,
2009. Three genomes from the phylum Acidobacteria provide
insight into the lifestyles of these microorganisms in soils.
Appl. Environ. Microbiol. 75 (7), 2046–2056.
Williams, M.M., Domingo, J., Meckes, M.C., Kelty, C.A., Rochon,
H.S., 2004. Phylogenetic diversity of drinking water bacteria in
a distribution system simulator. J. Appl. Microbiol. 96 (5),
954–964.
Xu, H., Roberts, N., Singleton, F.L., Attwell, R.W., Grimes, D.J.,
Colwell, R.R., 1982. Survival and viability of nonculturable
Escherichia coli and Vibrio cholerae in the estuarine and marine
environment. Microb. Ecol. 8 (4), 313–323.
Zhang, M., Liu, W., Nie, X., Li, C., Gu, J., Zhang, C., 2012. Molecular
analysis of bacterial communities in biofilms of a drinking
water clearwell. Microbes Environ. 27 (4), 443.
Zhang, D., Li, W., Gong, H., Zhang, L., Gong, X., Liu, B., 2013.
Evaluation of long term stability of seeded bacteria in a
bio-enhanced activated carbon filter used for treating
drinking water. Int. Biodeterior. Biodegrad. 85, 701–708 .
30 J O U R N A L O F E N V I R O N M E N T A L S C I E N C E S 5 1 ( 2 0 1 7 ) 2 1 – 3 0
1 out of 10
Related Documents

Your All-in-One AI-Powered Toolkit for Academic Success.
+13062052269
info@desklib.com
Available 24*7 on WhatsApp / Email
![[object Object]](/_next/static/media/star-bottom.7253800d.svg)
Unlock your academic potential
© 2024 | Zucol Services PVT LTD | All rights reserved.