Chediak-Higashi Syndrome: Phenotypes, Genotypes, Diagnosis
VerifiedAdded on 2023/01/18
|20
|6615
|87
Report
AI Summary
This report provides a comprehensive review of Chediak-Higashi Syndrome (CHS), an autosomal recessive disorder characterized by abnormally large lysosomes and other lysosome-related organelles. The report explores the genetic and molecular characteristics of CHS, with a particular emphasis on genotype-phenotype correlations, focusing on the LYST gene and its mutations. It details how the disease can be diagnosed, including the use of molecular techniques to identify the gene and the disease phenotype. Furthermore, the report discusses current molecular therapies for CHS and their mechanisms of action. The pathogenesis, diagnosis, and treatment options are discussed in detail, with a focus on the correlation between the genotype and the various phenotypes of the disease, including the accelerated phase and adult-onset forms. The report also examines the functional and structural overview of the LYST gene and the BEACH family of proteins, providing a thorough understanding of the disease's molecular basis and clinical manifestations. Overall, the report offers a detailed analysis of CHS, suitable for a short communication publication.

Running head: CHEDIAK-HIGASHI SYNDROME
CHEDIAK-HIGASHI SYNDROME
Name of the Student
Name of the University
Author Note
CHEDIAK-HIGASHI SYNDROME
Name of the Student
Name of the University
Author Note
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

1CHEDIAK-HIGASHI SYNDROME
Chediak-Higashi Syndrome (CHS) phenotype and Institution of Associating Genotypes
Acronyms
- CHS; Chediak Higashi Syndrome
- LRO; Lysosome-Related Organelles
- LYST; Lysosomal Trafficking Regulator Gene
- HLH; Haemophagocytic Lympho histocytosis
- PMN; Polymorphonuclear Leukocyte
- BEACH; Beige and Chediak Higashi
- PH; Pleckstrin Homology
- ARM; Armadilo
- HEAT; Huntington, EF3, PP2A and TOR1 protein domain
- IS; Immune Synapse
- CTL; Cytotoxic T-Cell
- NK; Natural Killer
- LCCA; Late Cortical Cerebellar Atrophy
- OPCA; Olivopontocerebellar Atrophy
- MRI; Magnetic Resonance Imaging
- APC; Antigen-presenting Cell
- EBV; Epstein Barr Virus
- UPD; UniparentalDisomy.
1
Chediak-Higashi Syndrome (CHS) phenotype and Institution of Associating Genotypes
Acronyms
- CHS; Chediak Higashi Syndrome
- LRO; Lysosome-Related Organelles
- LYST; Lysosomal Trafficking Regulator Gene
- HLH; Haemophagocytic Lympho histocytosis
- PMN; Polymorphonuclear Leukocyte
- BEACH; Beige and Chediak Higashi
- PH; Pleckstrin Homology
- ARM; Armadilo
- HEAT; Huntington, EF3, PP2A and TOR1 protein domain
- IS; Immune Synapse
- CTL; Cytotoxic T-Cell
- NK; Natural Killer
- LCCA; Late Cortical Cerebellar Atrophy
- OPCA; Olivopontocerebellar Atrophy
- MRI; Magnetic Resonance Imaging
- APC; Antigen-presenting Cell
- EBV; Epstein Barr Virus
- UPD; UniparentalDisomy.
1

2CHEDIAK-HIGASHI SYNDROME
Abstract
Chediak-Higashi Syndrome (CHS) is considered as an autosomal non-dominant syndrome, presented by the
development of aberrantly huge lysosomes and associated LROs that usually collect within peri nuclear
section of the cells. The inflated vesicle dimension includes the utility of above structures, resulting in
decreased bacterial devastation, incomplete albinism, neural problems, decreased coagulation and injury
repair. The patients usually surrender to repeated bacterial contaminations, or because of the principal organs
present in the body is subjected to the lymphoma-identical intrusion. Pathogenesis of disease in CHS is
connected with alteration actions expressing in wild or harsh kind human gene, termed as LYST, disturbing
the LYST gene expression. Although the biochemical activity of LYST is inadequate and has been gambled
to get involved in controlling the LROs and lysosome size by regulating either fusion events or vesicular
fission.
2
Abstract
Chediak-Higashi Syndrome (CHS) is considered as an autosomal non-dominant syndrome, presented by the
development of aberrantly huge lysosomes and associated LROs that usually collect within peri nuclear
section of the cells. The inflated vesicle dimension includes the utility of above structures, resulting in
decreased bacterial devastation, incomplete albinism, neural problems, decreased coagulation and injury
repair. The patients usually surrender to repeated bacterial contaminations, or because of the principal organs
present in the body is subjected to the lymphoma-identical intrusion. Pathogenesis of disease in CHS is
connected with alteration actions expressing in wild or harsh kind human gene, termed as LYST, disturbing
the LYST gene expression. Although the biochemical activity of LYST is inadequate and has been gambled
to get involved in controlling the LROs and lysosome size by regulating either fusion events or vesicular
fission.
2
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

3CHEDIAK-HIGASHI SYNDROME
Introduction
CHS is defined as an autosomal receding syndrome that is identified by flexible units of recurring
pyogenic contaminations, a predilection near minor bleeding incidents, neural dysfunction and
occulocutaneous albinism. Roughly 50 – 85% of patients suffering from CHS might feel an ‘enhanced
phase’ as portion of infection pathogenesis, connected with haemophagocytic lympho histocytosis (HLH)
that is frequently fatal if not diagnosed (4).
The genetic deficiency accountable for the above compulsive manifestations connected with CHS is
plotted to 1q42.3 chromosome, associating to the LYST gene in human. 422 kDa is total length of LYST
protein and includes highly preserved organization. Numerous ARM/HEAT α-helix replicates are situated
near the N-terminal area of protein, tracked by BEACH province and province of seven WD40 duplicates at
the domain of C-terminal (4).
The primary activity of LYST is focused to conjecture, and is considered to be connected with the
instruction of lysosome-associated organizations, comprising organelle dimension, secretion and fission. In
CHS, irregular genetic variations effect in massive cytoplasmic organelles like lysosomes, inclusion bodies
and melanosomes. Moreover, plasma membrane restoration mechanisms is subjected to resistance, in count
of the exocytic method engaged by lysosomes. A huge quantity of the figures, which permits the association
between phenotype and genotype in CHS that is extracted from the consumption of Lyst gene that is majorly
identical to the LYST human gene alternate (5).
Diagnosis
It is crucial to conduct a primary diagnosis of CHS. The usual age considered for CHS diagnosing is
nearly six years, although 25% of cases might be identified after age of 10 years. The analysis considered for
the disorder through prenatal phase is specified through the occurrence of productive phosphatase acid in the
lysosomes by amniotic liquid cells and foetus plasma leukocyte. The occurrence of large particles in the hair
tube is dangerous in diagnosis of syndrome through prenatal and antenatal phase (6). Hence, diagnosis of
CHS is entirely dependent on two different evaluations. First, the occurrence of huge positive peroxidase
particles in polymorph nuclear leukocytes (PMN) and secondly, genetic investigation of mutated gene for
LYST/CHS1 occurs in the principal chromosome. A dissimilar diagnostic research known as cytometric
fluorescence study that defines cellular surface particles and cell granules (6).
Various disorders sharing related symptoms of CHS exist. Though, they might be separated from CHS
by numerous remarks (6). Griscelli syndrome comprise of related symptoms as CHS comprising fractional
cutaneous-ophthalmic albinism, impaired phase through pancytopenia, cellular and humeral immune
insufficiency, hemophagocytosis, amplified serum triglyceride volume, reduced fibrinogens, haemorrhagic
syndrome and lessened plasma protein. Alteration develops in RAB27A and myo5a genes. Hermansky-
pudlak disease is another considered example where no huge granules is present. Platelet deposit is
exhausted related with bleeding. Respiratory fibrosis takes place. No neutrophil and defects occurs.
3
Introduction
CHS is defined as an autosomal receding syndrome that is identified by flexible units of recurring
pyogenic contaminations, a predilection near minor bleeding incidents, neural dysfunction and
occulocutaneous albinism. Roughly 50 – 85% of patients suffering from CHS might feel an ‘enhanced
phase’ as portion of infection pathogenesis, connected with haemophagocytic lympho histocytosis (HLH)
that is frequently fatal if not diagnosed (4).
The genetic deficiency accountable for the above compulsive manifestations connected with CHS is
plotted to 1q42.3 chromosome, associating to the LYST gene in human. 422 kDa is total length of LYST
protein and includes highly preserved organization. Numerous ARM/HEAT α-helix replicates are situated
near the N-terminal area of protein, tracked by BEACH province and province of seven WD40 duplicates at
the domain of C-terminal (4).
The primary activity of LYST is focused to conjecture, and is considered to be connected with the
instruction of lysosome-associated organizations, comprising organelle dimension, secretion and fission. In
CHS, irregular genetic variations effect in massive cytoplasmic organelles like lysosomes, inclusion bodies
and melanosomes. Moreover, plasma membrane restoration mechanisms is subjected to resistance, in count
of the exocytic method engaged by lysosomes. A huge quantity of the figures, which permits the association
between phenotype and genotype in CHS that is extracted from the consumption of Lyst gene that is majorly
identical to the LYST human gene alternate (5).
Diagnosis
It is crucial to conduct a primary diagnosis of CHS. The usual age considered for CHS diagnosing is
nearly six years, although 25% of cases might be identified after age of 10 years. The analysis considered for
the disorder through prenatal phase is specified through the occurrence of productive phosphatase acid in the
lysosomes by amniotic liquid cells and foetus plasma leukocyte. The occurrence of large particles in the hair
tube is dangerous in diagnosis of syndrome through prenatal and antenatal phase (6). Hence, diagnosis of
CHS is entirely dependent on two different evaluations. First, the occurrence of huge positive peroxidase
particles in polymorph nuclear leukocytes (PMN) and secondly, genetic investigation of mutated gene for
LYST/CHS1 occurs in the principal chromosome. A dissimilar diagnostic research known as cytometric
fluorescence study that defines cellular surface particles and cell granules (6).
Various disorders sharing related symptoms of CHS exist. Though, they might be separated from CHS
by numerous remarks (6). Griscelli syndrome comprise of related symptoms as CHS comprising fractional
cutaneous-ophthalmic albinism, impaired phase through pancytopenia, cellular and humeral immune
insufficiency, hemophagocytosis, amplified serum triglyceride volume, reduced fibrinogens, haemorrhagic
syndrome and lessened plasma protein. Alteration develops in RAB27A and myo5a genes. Hermansky-
pudlak disease is another considered example where no huge granules is present. Platelet deposit is
exhausted related with bleeding. Respiratory fibrosis takes place. No neutrophil and defects occurs.
3
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

4CHEDIAK-HIGASHI SYNDROME
Furthermore, Angelman and PraderWilli are two diseases comprised of hypopigmentation and absence of
ophthalmic albinism. Pyodermagangernosome, Warrensburg disease and Lazy leukocyte disease are
considered variance diagnosis. Mental retardation, neonatal Hyperphagia and hypogonadism are existent in
praderwilli condition. In Angelman disease, acute psychological retardation, ataxia, neonatal hypotonia,
microcephaly and incorrect laughter. In Warrensburg syndrome, neural crest deficiency, piebaldism,
congenital deafness, forelock hypopigmentation and extensive nasal root is observed (6).
Functional and Structural Overview
The LYST human gene conquers a huge area on the 1q42.3 chromosome, containing 55 exons and 222
kb length (7), as portrayed in Figure 1. The protein articulated by LYST gene is included in the family
BEACH (Beige and Chediak Higashi) of proteins. Every participants included in BEACH family has the
similar preserved arrangement encompassing C-terminal areas that comprise Pleckstrin homology (PH) and
sequence of WD-40 replicates monitored by BEACH province; N-terminal province remains highly vague
(7). The BEACH province is considered as the mark element within BEACH family, which is unique and
unusual. Large sections of the province either hidden internally or aid to remove hydrophobic core from the
present construction.
Figure 1: Chromosomal mapping of LYST gene present in chromosome 1
4
Furthermore, Angelman and PraderWilli are two diseases comprised of hypopigmentation and absence of
ophthalmic albinism. Pyodermagangernosome, Warrensburg disease and Lazy leukocyte disease are
considered variance diagnosis. Mental retardation, neonatal Hyperphagia and hypogonadism are existent in
praderwilli condition. In Angelman disease, acute psychological retardation, ataxia, neonatal hypotonia,
microcephaly and incorrect laughter. In Warrensburg syndrome, neural crest deficiency, piebaldism,
congenital deafness, forelock hypopigmentation and extensive nasal root is observed (6).
Functional and Structural Overview
The LYST human gene conquers a huge area on the 1q42.3 chromosome, containing 55 exons and 222
kb length (7), as portrayed in Figure 1. The protein articulated by LYST gene is included in the family
BEACH (Beige and Chediak Higashi) of proteins. Every participants included in BEACH family has the
similar preserved arrangement encompassing C-terminal areas that comprise Pleckstrin homology (PH) and
sequence of WD-40 replicates monitored by BEACH province; N-terminal province remains highly vague
(7). The BEACH province is considered as the mark element within BEACH family, which is unique and
unusual. Large sections of the province either hidden internally or aid to remove hydrophobic core from the
present construction.
Figure 1: Chromosomal mapping of LYST gene present in chromosome 1
4

5CHEDIAK-HIGASHI SYNDROME
Figure 2: The BEACH human family of proteins portrayed in respects to their province organisation.
The above provinces present at C-terminal area of BEACH family is wondered to be intricate in
compiling of protein associates, but their activity is still unidentified(7). Large statistics connecting to the
useful competences of LYST gene has been inferred through directing readings on other associates of the
descent, and the common agreement of them being related to receptor signalling, membrane dynamics and
vesicle trafficking (4).
Lately, it is exposed that lectin-like province is present in N-terminal area of BEACH family. The
lectin-alike province is ventured to initiate binding of oligosaccharide to protein trading, and also engage in
the practice of categorization beside the secretory way – this mainly relates to the essential constituents of
vesicular mixture technology (4).
Like the lectin-like province that is preserved feature in the BEACH proteins descent, the N’ end area
of LYST protein comprises numerous ARM (Armadilo)/HEAT (protein province establish in Huntingtin,
EF3, PP2A, and TOR1) α-helix recurrence motifs, precise to the present protein. Therefore, the synergistic
act of the LYST protein subunits permit them to be accountable for fission and union actions that effect the
complete dimension of the cellular arrangements, with its effect not exclusively limited to lysosomal
constructions, but also influences several alike organelles (5).
LYST is intricate in either vesicle fission or fusion with a serious part in the endolysosomal biogenesis
which controls the terminal development of secreted lysosomes
The researcher have dedicated on the trademark distended cytoplasmic organelles existent in
practically every granulated cells (1) of the sufferers with CHS. The above organelles are suggested to
establish a combination of lysosomal and endosomal structures (11), which has identical degradation ability
as compared to lysosomal (8). Though an agreement on the process of these huge organelles formation has
5
Figure 2: The BEACH human family of proteins portrayed in respects to their province organisation.
The above provinces present at C-terminal area of BEACH family is wondered to be intricate in
compiling of protein associates, but their activity is still unidentified(7). Large statistics connecting to the
useful competences of LYST gene has been inferred through directing readings on other associates of the
descent, and the common agreement of them being related to receptor signalling, membrane dynamics and
vesicle trafficking (4).
Lately, it is exposed that lectin-like province is present in N-terminal area of BEACH family. The
lectin-alike province is ventured to initiate binding of oligosaccharide to protein trading, and also engage in
the practice of categorization beside the secretory way – this mainly relates to the essential constituents of
vesicular mixture technology (4).
Like the lectin-like province that is preserved feature in the BEACH proteins descent, the N’ end area
of LYST protein comprises numerous ARM (Armadilo)/HEAT (protein province establish in Huntingtin,
EF3, PP2A, and TOR1) α-helix recurrence motifs, precise to the present protein. Therefore, the synergistic
act of the LYST protein subunits permit them to be accountable for fission and union actions that effect the
complete dimension of the cellular arrangements, with its effect not exclusively limited to lysosomal
constructions, but also influences several alike organelles (5).
LYST is intricate in either vesicle fission or fusion with a serious part in the endolysosomal biogenesis
which controls the terminal development of secreted lysosomes
The researcher have dedicated on the trademark distended cytoplasmic organelles existent in
practically every granulated cells (1) of the sufferers with CHS. The above organelles are suggested to
establish a combination of lysosomal and endosomal structures (11), which has identical degradation ability
as compared to lysosomal (8). Though an agreement on the process of these huge organelles formation has
5
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

6CHEDIAK-HIGASHI SYNDROME
not reached, considering the investigation data it was exposed that LYST has a crucial character in the
endolysosomal biogenesis that controls the end development of secretive lysosomes (11). Patients with CHS
have enlarged cytotoxic granules and miscarry to issue their material at the immune synapse (IS), in spite of
a usual polarization during initiation (1). Therefore, LYST is included in decreased fission or amplified
vesicle fusion (1, 3, 16, 17); the best auspicious prototypes is in support of former (18, 19).
Various readings have projected that manifestation of beige protein can be harmfully control
prelysosomal vesicles or homotypic endosome union, the absenteeism would upsurge combination of
vesicles, ensuing in huge lysosomes (17). Therefore, the appearance of medical indicators connected with the
syndrome is highly connected to the lysosome capacity to combine with organelles (4).
Future study established that LYST protein is
mandatory for the end development of perforin-
comprising particles into secretive cytotoxic particles
(11). Autonomous size, perforin-comprising particles
in CHS. CTLs exhibited an extreme, docked-alike
phenotype at IS but was unable to fuse and degranulate
(11). The identified lytic particle exocytosis effectors
(see Figure 4) are localized in late endosomal sections
and reutilizing in standard CTLs were misguided along
the distended lysosomal organelle present in CHS
CTLs (11). Misguidance of the effectors will result in
perforin-facilitated cytotoxic impairment activity in
CTLs (1) and natural killer (NK) cells, leading to
immune insufficiency and development to HLH.
Genotype–Phenotype association of CHS
Researchers have determined that a resilient phenotype-genotype association is present for CHS with a
characteristic phenotypically heterogeneity. This heterogeneity is specifically dominant in a varying manner
of alteration in the similar CHS1/LYST gene that is evident as considerably changed phenotype (8).
Usually, the complex CHS, accompanied with early inception of the enhanced stage and connected
HLH in infancy, is credited to frame shift, nonsense, or intertwine site alterations leading to vague LYST
protein, whereas the minor type of CHS, accompanied with later inception in middle age and missing the
development of HLH that is accredited to one missense alteration, causing partial function protein (7). The
mentioned varying kinds of alterations connected to LYST gene assembly are portrayed as Figure 3.
Figure 4: the lysosomal–endosomal device is accountable
for intracellular breakdown of internally and externally
produced macromolecules.
6
not reached, considering the investigation data it was exposed that LYST has a crucial character in the
endolysosomal biogenesis that controls the end development of secretive lysosomes (11). Patients with CHS
have enlarged cytotoxic granules and miscarry to issue their material at the immune synapse (IS), in spite of
a usual polarization during initiation (1). Therefore, LYST is included in decreased fission or amplified
vesicle fusion (1, 3, 16, 17); the best auspicious prototypes is in support of former (18, 19).
Various readings have projected that manifestation of beige protein can be harmfully control
prelysosomal vesicles or homotypic endosome union, the absenteeism would upsurge combination of
vesicles, ensuing in huge lysosomes (17). Therefore, the appearance of medical indicators connected with the
syndrome is highly connected to the lysosome capacity to combine with organelles (4).
Future study established that LYST protein is
mandatory for the end development of perforin-
comprising particles into secretive cytotoxic particles
(11). Autonomous size, perforin-comprising particles
in CHS. CTLs exhibited an extreme, docked-alike
phenotype at IS but was unable to fuse and degranulate
(11). The identified lytic particle exocytosis effectors
(see Figure 4) are localized in late endosomal sections
and reutilizing in standard CTLs were misguided along
the distended lysosomal organelle present in CHS
CTLs (11). Misguidance of the effectors will result in
perforin-facilitated cytotoxic impairment activity in
CTLs (1) and natural killer (NK) cells, leading to
immune insufficiency and development to HLH.
Genotype–Phenotype association of CHS
Researchers have determined that a resilient phenotype-genotype association is present for CHS with a
characteristic phenotypically heterogeneity. This heterogeneity is specifically dominant in a varying manner
of alteration in the similar CHS1/LYST gene that is evident as considerably changed phenotype (8).
Usually, the complex CHS, accompanied with early inception of the enhanced stage and connected
HLH in infancy, is credited to frame shift, nonsense, or intertwine site alterations leading to vague LYST
protein, whereas the minor type of CHS, accompanied with later inception in middle age and missing the
development of HLH that is accredited to one missense alteration, causing partial function protein (7). The
mentioned varying kinds of alterations connected to LYST gene assembly are portrayed as Figure 3.
Figure 4: the lysosomal–endosomal device is accountable
for intracellular breakdown of internally and externally
produced macromolecules.
6
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

7CHEDIAK-HIGASHI SYNDROME
Figure 3: The LYST gene in human portrayed in respects to its construction and connected mutational
actions.
Therefore, during the later-inception of CHS it is commonly accredited to similar missense alterations,
protein truncating alterations is recognized not individually in adult type of syndrome, also included in
juvenile variation. Hence, onset interval in people with homozygous shortening alterations are credited to
both genetic and environmental factors (13). Conversely, the recognition of the genuine molecular outcome
during molecular variation is essential to begin a phenotype-genotype association in sufferers owning a dual-
allelic missense alteration (7). The institution of the phenotype-genotype association in respects to CHS
permits for the investigation of the way that give alternation actions distress the appearance of the harsh
LYST gene kind.
Alteration of the CHS1/LYST Gene
By discovering the genotypic centre consisting of phenotypic manifestation of CHS, there is
possibility to extravagant on the phenotypically genetic related with the syndrome (7). The CHS1/LYST gene
encrypts for the lysosomal operating protein, the method of subcellular operation in CHS comprise by the
harm of gene manifestation that has been matter to numerous readings, in an effort to recognize the method
through that procedure may happen (6).
Immune inadequacy frequently demonstrates in those personalities with the complex alternative of the
syndrome, with a tendency to an enhanced stage because of transmissible procedure or bleeding event that
often leads to demise of an individual (10).
While the complexity of CHS have been conventionally separated by phenotypic manifestation in
respects to the syndrome pathogenesis process, CHS is separated on the basis of genotype, endorsing a more
7
Figure 3: The LYST gene in human portrayed in respects to its construction and connected mutational
actions.
Therefore, during the later-inception of CHS it is commonly accredited to similar missense alterations,
protein truncating alterations is recognized not individually in adult type of syndrome, also included in
juvenile variation. Hence, onset interval in people with homozygous shortening alterations are credited to
both genetic and environmental factors (13). Conversely, the recognition of the genuine molecular outcome
during molecular variation is essential to begin a phenotype-genotype association in sufferers owning a dual-
allelic missense alteration (7). The institution of the phenotype-genotype association in respects to CHS
permits for the investigation of the way that give alternation actions distress the appearance of the harsh
LYST gene kind.
Alteration of the CHS1/LYST Gene
By discovering the genotypic centre consisting of phenotypic manifestation of CHS, there is
possibility to extravagant on the phenotypically genetic related with the syndrome (7). The CHS1/LYST gene
encrypts for the lysosomal operating protein, the method of subcellular operation in CHS comprise by the
harm of gene manifestation that has been matter to numerous readings, in an effort to recognize the method
through that procedure may happen (6).
Immune inadequacy frequently demonstrates in those personalities with the complex alternative of the
syndrome, with a tendency to an enhanced stage because of transmissible procedure or bleeding event that
often leads to demise of an individual (10).
While the complexity of CHS have been conventionally separated by phenotypic manifestation in
respects to the syndrome pathogenesis process, CHS is separated on the basis of genotype, endorsing a more
7

8CHEDIAK-HIGASHI SYNDROME
complete analysis. Mainly, this genotypic separation have been recognized through consumption of a CHS
mouse model (7).
A mouse comprising of different “beige” covering colour is used as an ideal in CHS, which was
considered as the chief occurrence for the gene accountable to the disease was recognized (15). 87.9%
homologous behaviour is seen in the LYST/CHS1 gene within murine gene in CHS – denoted as lyst
considered as the mouse prototype of human beige (7).
The examination genotypic alteration degree and the method through which it associates to syndrome
pathogenesis like a phenotypic manifestation permits for separation of CHS, relied on syndrome
rigorousness (16). By creating the phenotype-genotype association in respects to the manifestation of the
harsh kind, the essential foundation is delivered to examine the phenotype-genotype association in the
insignificantly altered allelic set and harshly altered allelic set (7).
Expression, specific cell type, regulation
From the result of the relation among CHS and LYST gene, gene plotting was executed by the
connection readings of CHS. The result exhibited that the CHS gene dwelling might be in 1q distal as the
similar condition in mouse towards CHS offered association to nidogen gene (131390) positioned on 1q
human. The D1S235 indicator achieved the highest lod value (5.38 at theta = 0) by consuming the haplotype
inspection to illustrate D1S163 into the centromere neighbouring indicator and D1S2680 into the telomere
neighbouring indicator (7).
Evaluation of similar coloration alterations in human, mouse and further model creatures result in
arrangement of the current evidence on all coloration genes copied from mouse or human, and noted on three
careful structures (18). A sole family of gene is identified from the connection between melanosomes and
lysosomes, which include LYST and HPS (203300). A robust interruption takes place in antigen exhibition
and peptide stacking onto chief histocompatibility complex (HTC) class II particles in CHS cells of the
sufferers. Categorization of endosomal inhabitant proteins is reliant on the product of CHS1 gene’s for
converting in late endosomes by a technique that includes microtubules (7).
Minor Genotype-phenotype association
Approximately, 15% of CHS sufferers developed an insignificant form of the disorder exhibiting a
different phenotype, while 85% advance a severe definitive form of the syndrome during and post birth (21).
Presently, explanation of 63 CHS1/LYST alterations that comprise 4 acceptor splice positions, 19 deletions,
31 substitutions and 9 insertions (20 non-senses and 11 missense) (19). A noteworthy up-front association
for phenotype and genotype of the disorder is planned: primary reports displays frame shift, splice site and
non-sense mutations resulting in deficiency of CHS1/LYST protein and those linked to acute CHS in infant,
while minor CHS in youngsters and grownups comprise one missense alteration that encrypts a protein
working incompletely (7).
8
complete analysis. Mainly, this genotypic separation have been recognized through consumption of a CHS
mouse model (7).
A mouse comprising of different “beige” covering colour is used as an ideal in CHS, which was
considered as the chief occurrence for the gene accountable to the disease was recognized (15). 87.9%
homologous behaviour is seen in the LYST/CHS1 gene within murine gene in CHS – denoted as lyst
considered as the mouse prototype of human beige (7).
The examination genotypic alteration degree and the method through which it associates to syndrome
pathogenesis like a phenotypic manifestation permits for separation of CHS, relied on syndrome
rigorousness (16). By creating the phenotype-genotype association in respects to the manifestation of the
harsh kind, the essential foundation is delivered to examine the phenotype-genotype association in the
insignificantly altered allelic set and harshly altered allelic set (7).
Expression, specific cell type, regulation
From the result of the relation among CHS and LYST gene, gene plotting was executed by the
connection readings of CHS. The result exhibited that the CHS gene dwelling might be in 1q distal as the
similar condition in mouse towards CHS offered association to nidogen gene (131390) positioned on 1q
human. The D1S235 indicator achieved the highest lod value (5.38 at theta = 0) by consuming the haplotype
inspection to illustrate D1S163 into the centromere neighbouring indicator and D1S2680 into the telomere
neighbouring indicator (7).
Evaluation of similar coloration alterations in human, mouse and further model creatures result in
arrangement of the current evidence on all coloration genes copied from mouse or human, and noted on three
careful structures (18). A sole family of gene is identified from the connection between melanosomes and
lysosomes, which include LYST and HPS (203300). A robust interruption takes place in antigen exhibition
and peptide stacking onto chief histocompatibility complex (HTC) class II particles in CHS cells of the
sufferers. Categorization of endosomal inhabitant proteins is reliant on the product of CHS1 gene’s for
converting in late endosomes by a technique that includes microtubules (7).
Minor Genotype-phenotype association
Approximately, 15% of CHS sufferers developed an insignificant form of the disorder exhibiting a
different phenotype, while 85% advance a severe definitive form of the syndrome during and post birth (21).
Presently, explanation of 63 CHS1/LYST alterations that comprise 4 acceptor splice positions, 19 deletions,
31 substitutions and 9 insertions (20 non-senses and 11 missense) (19). A noteworthy up-front association
for phenotype and genotype of the disorder is planned: primary reports displays frame shift, splice site and
non-sense mutations resulting in deficiency of CHS1/LYST protein and those linked to acute CHS in infant,
while minor CHS in youngsters and grownups comprise one missense alteration that encrypts a protein
working incompletely (7).
8
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

9CHEDIAK-HIGASHI SYNDROME
Lysosomal operating controller protein is determined by LYST gene that is included in monitoring the
dimension of vesicles therefore conveying them into the cells. Melanosomes denote to lysosome-alike
vesicles, which occur in melanocytes, whereas the storage and synthesis of melanin previous to activity to
neighbouring keratinocytes (22). Efficient readings including numerous provinces of LYST including the
recognized associating LYST companions intensely exhibit the activity of LYST, in inaugurating the
organelle dimension through regulation of membrane separation actions. The trademark in CHS is addition
of vast bodies in the granulated cells, vast lysosomes together with huge melanosomes. In America, the
clinical indicators of CHS almost ignored the CHS in humans, those inclined to progress a
lymphoproliferative disease in the augmented phase. Separately from the distinction, the idea that CHS
indicators in mink is not simply identified as linked from the humans may be accredited to the opinion that
contamination revelation is low as the mink farmhouses are typically sheltered from syndromes (22).
CHS1 protein action is principally unidentified; though, there is exhibition of distended lysosomes
along with lysosome-focused organelles in all types of cells in CHS sufferers. Modern functional
investigation on numerous provinces of CHS1 and its cooperating associates with v-SNARES intensely
specify CHS1 character in identifying the organelle dimension over instruction of membrane separation and
separation actions (23).
Numerous clinical phenotypes of CHS is correlated with molecular genotyping as compared to
missense alterations are related to minor, late-inception CHS having sluggish advanced neurological
imperfection, or the youthful type that has contaminations but deficit of haemophagocytic lympohistiocytosis
(23).
Late-inception neural dysfunction
A neurodegenerative advanced disorder enclosing exceptional pigmentary deficiencies and missing
subclinical invulnerable deficiencies known as late-inception CHS. The mentioned CHS holds for 10-15%
for all CHS patients; though, the number can be increased (20). Neurologic indications likely to fluctuate, but
occasionally encompass of Parkinsonism, ataxia, peripheral neuropathy and intellectual reduction. The
people with genotype including the mentioned phenotypes resulted in four genotypes, which result in
LYST/CHS; consisting missense alterations (24). CHS murine models comprising homozygous alterations of
missense in the LYST exhibit various neurologic phenotypes like inferior motor functionality marks equated
to standards, with assortment of huge lysosomes within cellular neurons associated with intracytoplasmic
combination in Purkinje cells consisting of cerebellum and motor cortex. Through autopsy, the adult patient
with CHS exhibit a failure in neurons that included the fragile olivary nuclei together with the cerebellar
cortex, joined with a whole dispersal that is similar to OPCA (olivopontocerebellar atrophy) and LCCA (late
cortical cerebellar atrophy) without any pontine participation (25).
Three grownup siblings was utilized to demonstrate a separate type of Chediak-Higashi disease acting
with neurodegenerative development disorder and partaking minor immunological imperfections, associated
9
Lysosomal operating controller protein is determined by LYST gene that is included in monitoring the
dimension of vesicles therefore conveying them into the cells. Melanosomes denote to lysosome-alike
vesicles, which occur in melanocytes, whereas the storage and synthesis of melanin previous to activity to
neighbouring keratinocytes (22). Efficient readings including numerous provinces of LYST including the
recognized associating LYST companions intensely exhibit the activity of LYST, in inaugurating the
organelle dimension through regulation of membrane separation actions. The trademark in CHS is addition
of vast bodies in the granulated cells, vast lysosomes together with huge melanosomes. In America, the
clinical indicators of CHS almost ignored the CHS in humans, those inclined to progress a
lymphoproliferative disease in the augmented phase. Separately from the distinction, the idea that CHS
indicators in mink is not simply identified as linked from the humans may be accredited to the opinion that
contamination revelation is low as the mink farmhouses are typically sheltered from syndromes (22).
CHS1 protein action is principally unidentified; though, there is exhibition of distended lysosomes
along with lysosome-focused organelles in all types of cells in CHS sufferers. Modern functional
investigation on numerous provinces of CHS1 and its cooperating associates with v-SNARES intensely
specify CHS1 character in identifying the organelle dimension over instruction of membrane separation and
separation actions (23).
Numerous clinical phenotypes of CHS is correlated with molecular genotyping as compared to
missense alterations are related to minor, late-inception CHS having sluggish advanced neurological
imperfection, or the youthful type that has contaminations but deficit of haemophagocytic lympohistiocytosis
(23).
Late-inception neural dysfunction
A neurodegenerative advanced disorder enclosing exceptional pigmentary deficiencies and missing
subclinical invulnerable deficiencies known as late-inception CHS. The mentioned CHS holds for 10-15%
for all CHS patients; though, the number can be increased (20). Neurologic indications likely to fluctuate, but
occasionally encompass of Parkinsonism, ataxia, peripheral neuropathy and intellectual reduction. The
people with genotype including the mentioned phenotypes resulted in four genotypes, which result in
LYST/CHS; consisting missense alterations (24). CHS murine models comprising homozygous alterations of
missense in the LYST exhibit various neurologic phenotypes like inferior motor functionality marks equated
to standards, with assortment of huge lysosomes within cellular neurons associated with intracytoplasmic
combination in Purkinje cells consisting of cerebellum and motor cortex. Through autopsy, the adult patient
with CHS exhibit a failure in neurons that included the fragile olivary nuclei together with the cerebellar
cortex, joined with a whole dispersal that is similar to OPCA (olivopontocerebellar atrophy) and LCCA (late
cortical cerebellar atrophy) without any pontine participation (25).
Three grownup siblings was utilized to demonstrate a separate type of Chediak-Higashi disease acting
with neurodegenerative development disorder and partaking minor immunological imperfections, associated
9
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

10CHEDIAK-HIGASHI SYNDROME
with unusual coloration (24). The patients displayed neuropsychological insufficiencies like attention-deficit
behaviours and learning problems that may have significantly preceded the commencement of free
neurologic signs. Additionally, traditional CHS is related to learning difficulties in lawbreakers, and further
to advanced, dispersed white matter imperfections on MRI joined with neurologic decline and amplified
worsening in immunity or resistance. The family encompasses CHS phenotype through contributing more
description for the syndromes connected with the CNS of the patients. Irregular CHS is considered
phenotypically dissimilar and must be taken in account during growth of neurodegenerative disorder escorted
by pigmentary deficiencies in young grown-ups. The syndrome can be separated easily by distinguishing
outcomes from marginal blood slur and hair selection, even if these are not common as related to standard
CHS phenotypes (24).
The early-inception phenotype is categorized by the inclination to create an ‘enhanced phase’
The early-inception phenotype is categorized by the inclination to create an ‘enhanced phase’, a lymph
proliferative penetration of the reticuloendothelial system and bone marrow that relates to the scientific
depiction of HLH (1, 25, 26). HLH might take place immediately after or few years of birth (27) and is
described by an extremely enthused, but unsuccessful, immune reaction to antigens, causing a dangerous
cytokine hurricane and inflammatory response (1). The granule-reliant cytotoxic path of CTLs and NK cells
is the crucial equipment in the sanction of intracellular and viral bacterial contaminations, including the
tumour growth prevention (11). This comprises cytotoxic particle biogenesis and end development, operating
to IS, priming, cutting, and blending with plasma membrane (PM) to adapt secretion capability (see Figure 5)
(11). In a fit person, the above cytotoxic particles comprising perforin, supplemented by granzymes, which
facilitate apoptotic demise of the target cells tailed by directive of immune reaction (28). HLH is connected
with an overexcited adaptive immune structure, possibly resultant due to disaster of stimulated NK cells and
CTLs to remove antigen-presenting cells (APCs), therefore, miscarry to stop the immune reaction. This
effects from the issue of perforin or unsuccessful biogenesis, the crucial cytotoxic moderator (29).
Figure 5: Schematic illustration of HLHs with their faulty genes.
10
with unusual coloration (24). The patients displayed neuropsychological insufficiencies like attention-deficit
behaviours and learning problems that may have significantly preceded the commencement of free
neurologic signs. Additionally, traditional CHS is related to learning difficulties in lawbreakers, and further
to advanced, dispersed white matter imperfections on MRI joined with neurologic decline and amplified
worsening in immunity or resistance. The family encompasses CHS phenotype through contributing more
description for the syndromes connected with the CNS of the patients. Irregular CHS is considered
phenotypically dissimilar and must be taken in account during growth of neurodegenerative disorder escorted
by pigmentary deficiencies in young grown-ups. The syndrome can be separated easily by distinguishing
outcomes from marginal blood slur and hair selection, even if these are not common as related to standard
CHS phenotypes (24).
The early-inception phenotype is categorized by the inclination to create an ‘enhanced phase’
The early-inception phenotype is categorized by the inclination to create an ‘enhanced phase’, a lymph
proliferative penetration of the reticuloendothelial system and bone marrow that relates to the scientific
depiction of HLH (1, 25, 26). HLH might take place immediately after or few years of birth (27) and is
described by an extremely enthused, but unsuccessful, immune reaction to antigens, causing a dangerous
cytokine hurricane and inflammatory response (1). The granule-reliant cytotoxic path of CTLs and NK cells
is the crucial equipment in the sanction of intracellular and viral bacterial contaminations, including the
tumour growth prevention (11). This comprises cytotoxic particle biogenesis and end development, operating
to IS, priming, cutting, and blending with plasma membrane (PM) to adapt secretion capability (see Figure 5)
(11). In a fit person, the above cytotoxic particles comprising perforin, supplemented by granzymes, which
facilitate apoptotic demise of the target cells tailed by directive of immune reaction (28). HLH is connected
with an overexcited adaptive immune structure, possibly resultant due to disaster of stimulated NK cells and
CTLs to remove antigen-presenting cells (APCs), therefore, miscarry to stop the immune reaction. This
effects from the issue of perforin or unsuccessful biogenesis, the crucial cytotoxic moderator (29).
Figure 5: Schematic illustration of HLHs with their faulty genes.
10
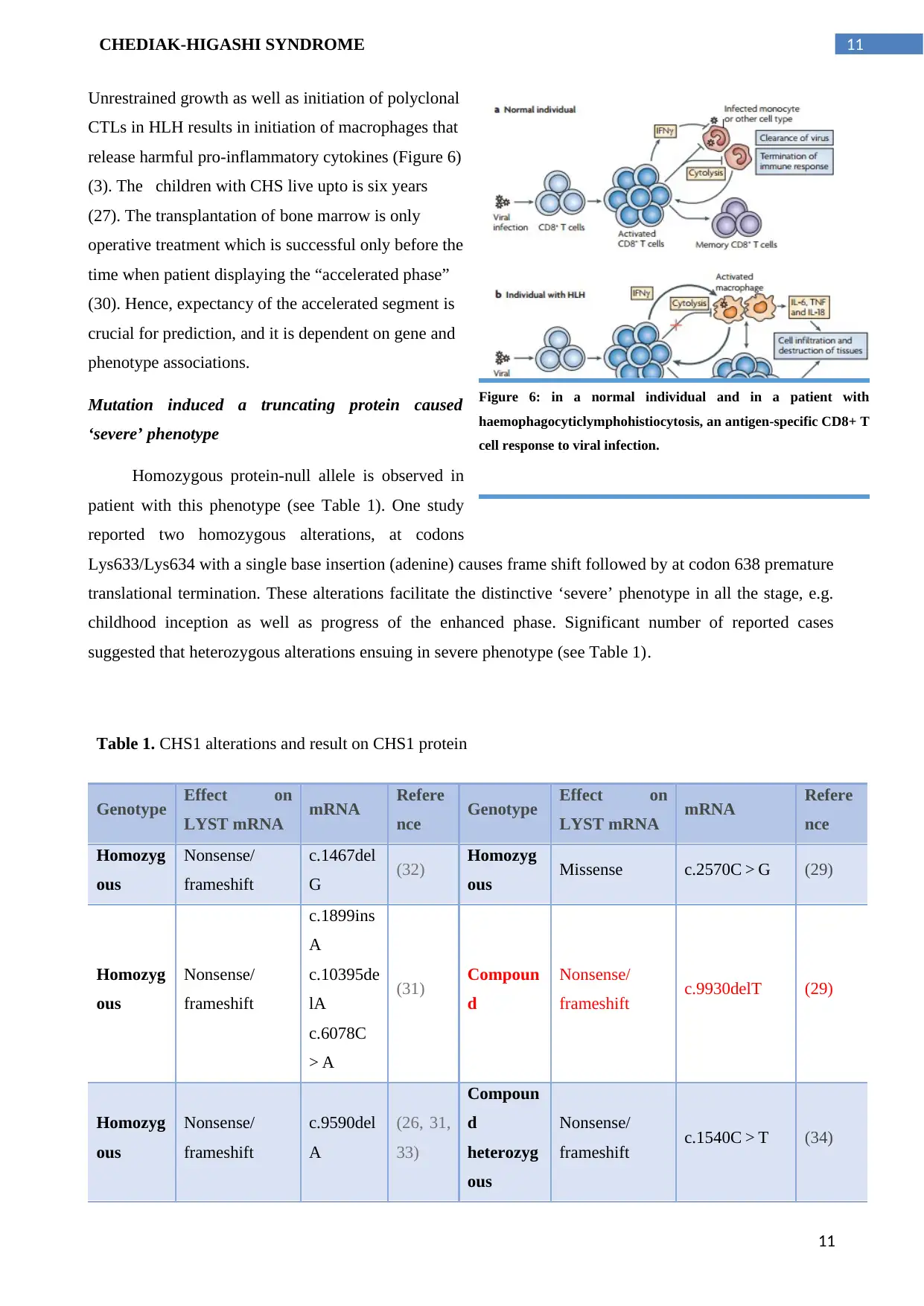
11CHEDIAK-HIGASHI SYNDROME
Unrestrained growth as well as initiation of polyclonal
CTLs in HLH results in initiation of macrophages that
release harmful pro-inflammatory cytokines (Figure 6)
(3). The children with CHS live upto is six years
(27). The transplantation of bone marrow is only
operative treatment which is successful only before the
time when patient displaying the “accelerated phase”
(30). Hence, expectancy of the accelerated segment is
crucial for prediction, and it is dependent on gene and
phenotype associations.
Mutation induced a truncating protein caused
‘severe’ phenotype
Homozygous protein-null allele is observed in
patient with this phenotype (see Table 1). One study
reported two homozygous alterations, at codons
Lys633/Lys634 with a single base insertion (adenine) causes frame shift followed by at codon 638 premature
translational termination. These alterations facilitate the distinctive ‘severe’ phenotype in all the stage, e.g.
childhood inception as well as progress of the enhanced phase. Significant number of reported cases
suggested that heterozygous alterations ensuing in severe phenotype (see Table 1).
Table 1. CHS1 alterations and result on CHS1 protein
Genotype Effect on
LYST mRNA mRNA Refere
nce Genotype Effect on
LYST mRNA mRNA Refere
nce
Homozyg
ous
Nonsense/
frameshift
c.1467del
G (32) Homozyg
ous Missense c.2570C > G (29)
Homozyg
ous
Nonsense/
frameshift
c.1899ins
A
c.10395de
lA
c.6078C
> A
(31) Compoun
d
Nonsense/
frameshift c.9930delT (29)
Homozyg
ous
Nonsense/
frameshift
c.9590del
A
(26, 31,
33)
Compoun
d
heterozyg
ous
Nonsense/
frameshift c.1540C > T (34)
Figure 6: in a normal individual and in a patient with
haemophagocyticlymphohistiocytosis, an antigen-specific CD8+ T
cell response to viral infection.
11
Unrestrained growth as well as initiation of polyclonal
CTLs in HLH results in initiation of macrophages that
release harmful pro-inflammatory cytokines (Figure 6)
(3). The children with CHS live upto is six years
(27). The transplantation of bone marrow is only
operative treatment which is successful only before the
time when patient displaying the “accelerated phase”
(30). Hence, expectancy of the accelerated segment is
crucial for prediction, and it is dependent on gene and
phenotype associations.
Mutation induced a truncating protein caused
‘severe’ phenotype
Homozygous protein-null allele is observed in
patient with this phenotype (see Table 1). One study
reported two homozygous alterations, at codons
Lys633/Lys634 with a single base insertion (adenine) causes frame shift followed by at codon 638 premature
translational termination. These alterations facilitate the distinctive ‘severe’ phenotype in all the stage, e.g.
childhood inception as well as progress of the enhanced phase. Significant number of reported cases
suggested that heterozygous alterations ensuing in severe phenotype (see Table 1).
Table 1. CHS1 alterations and result on CHS1 protein
Genotype Effect on
LYST mRNA mRNA Refere
nce Genotype Effect on
LYST mRNA mRNA Refere
nce
Homozyg
ous
Nonsense/
frameshift
c.1467del
G (32) Homozyg
ous Missense c.2570C > G (29)
Homozyg
ous
Nonsense/
frameshift
c.1899ins
A
c.10395de
lA
c.6078C
> A
(31) Compoun
d
Nonsense/
frameshift c.9930delT (29)
Homozyg
ous
Nonsense/
frameshift
c.9590del
A
(26, 31,
33)
Compoun
d
heterozyg
ous
Nonsense/
frameshift c.1540C > T (34)
Figure 6: in a normal individual and in a patient with
haemophagocyticlymphohistiocytosis, an antigen-specific CD8+ T
cell response to viral infection.
11
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide
1 out of 20
Your All-in-One AI-Powered Toolkit for Academic Success.
+13062052269
info@desklib.com
Available 24*7 on WhatsApp / Email
![[object Object]](/_next/static/media/star-bottom.7253800d.svg)
Unlock your academic potential
Copyright © 2020–2026 A2Z Services. All Rights Reserved. Developed and managed by ZUCOL.