Cleanroom Design, Validation, and Maintenance Report
VerifiedAdded on 2023/06/04
|34
|7315
|299
Report
AI Summary
This report provides a comprehensive overview of cleanroom validation within the context of biotechnology drug manufacturing, specifically focusing on the production of Remicade. It begins by exploring the fundamental principles of cleanroom design, emphasizing the importance of protecting the product from contamination and the strategic considerations for layout, including changing rooms and material distribution. The report then delves into the critical steps of cleanroom validation, detailing the Design Qualification (DQ), Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ) phases, alongside cleanroom certification and monitoring protocols. Furthermore, it outlines the essential procedures for maintaining a validated cleanroom state, including maintenance schedules, cleaning instructions, and the significance of cleanroom clothing and general regulations. The report underscores the crucial role of air quality, particle measurement, and the application of standards like ISO 14644 to ensure a controlled and sterile environment conducive to the production of high-quality biotechnology drugs. It addresses the challenges of contamination control from various sources, including personnel, equipment, and processes, and emphasizes the need for systematic engineering designs and complex technological tools to achieve and sustain the required air quality parameters. The report highlights that the overall goal is to reduce risk of contamination, while concurrently developing a facility which operates without adding constraints to the unit cost of the product and is economical to design.

CLEANROOM VALIDATION 1
Cleanroom Validation
Student Name
Institutional Affiliation
Cleanroom Validation
Student Name
Institutional Affiliation
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

CLEANROOM VALIDATION 2
Table of Contents
1 Introduction..............................................................................................................................3
2 Designing Cleanrooms.............................................................................................................4
2.1 Strategy and Purpose of design.........................................................................................4
2.2 Cleanroom Layout.............................................................................................................6
2.3 Changing room..................................................................................................................9
2.4 Material distribution technology.....................................................................................10
2.4.1 Air supply................................................................................................................10
2.4.2 Air distribution.........................................................................................................11
2.4.3 Air Filtration and supply..........................................................................................15
2.4.4 Construction Materials.............................................................................................16
2.4.5 Operating procedures...............................................................................................17
3 Cleanroom Validation............................................................................................................17
3.1 Lifecycle of Cleanroom Validation.................................................................................18
3.1.1 DQ (Design Qualification)......................................................................................19
3.1.2 IQ (Installation Qualification).................................................................................19
3.1.3 OQ (Operation Qualification)..................................................................................21
3.1.4 PQ (Performance Qualification)..............................................................................22
3.2 Cleanroom Certification..................................................................................................23
3.3 Monitor and Control........................................................................................................23
4 Maintenance and Control of Cleanroom Areas......................................................................24
5 Cleanroom Clothing...............................................................................................................26
6 Cleaning instructions..............................................................................................................27
6.1 General regulations of a cleanroom................................................................................27
6.2 Airborne Particle Measurement......................................................................................28
7 Conclusion..............................................................................................................................28
Reference List................................................................................................................................30
Table of Contents
1 Introduction..............................................................................................................................3
2 Designing Cleanrooms.............................................................................................................4
2.1 Strategy and Purpose of design.........................................................................................4
2.2 Cleanroom Layout.............................................................................................................6
2.3 Changing room..................................................................................................................9
2.4 Material distribution technology.....................................................................................10
2.4.1 Air supply................................................................................................................10
2.4.2 Air distribution.........................................................................................................11
2.4.3 Air Filtration and supply..........................................................................................15
2.4.4 Construction Materials.............................................................................................16
2.4.5 Operating procedures...............................................................................................17
3 Cleanroom Validation............................................................................................................17
3.1 Lifecycle of Cleanroom Validation.................................................................................18
3.1.1 DQ (Design Qualification)......................................................................................19
3.1.2 IQ (Installation Qualification).................................................................................19
3.1.3 OQ (Operation Qualification)..................................................................................21
3.1.4 PQ (Performance Qualification)..............................................................................22
3.2 Cleanroom Certification..................................................................................................23
3.3 Monitor and Control........................................................................................................23
4 Maintenance and Control of Cleanroom Areas......................................................................24
5 Cleanroom Clothing...............................................................................................................26
6 Cleaning instructions..............................................................................................................27
6.1 General regulations of a cleanroom................................................................................27
6.2 Airborne Particle Measurement......................................................................................28
7 Conclusion..............................................................................................................................28
Reference List................................................................................................................................30

CLEANROOM VALIDATION 3
1 Introduction
Presently cleanrooms are utilized in several industries such as biotechnology, pharmaceuticals,
defense industry, microelectronics and nanotechnology. Cleanrooms may vary from small to
large multilevel structures with big serviced utilities and equipment. Cleanroom is a managed
placement where various goods are produced and airborne particles concentration is managed to
particularized limits. As such, the killing process of contaminants of ultrafines need to be
controlled. Contaminations are created by processes, equipment, people and facilities. They must
be constantly eliminated from the air. The air cleanliness level in a room should be controlled by
standards. ISO 14644 is the most regularly applied standard (Brahm, 2012).
The primary cleanroom function is to safeguard the produced product from contamination. In
manufacturing of a biotechnology drug, manufacturer economic survival relies on the protection
of the manufactured product. Therefore, a potential contamination source should be identified,
including the working environment. Supply air quality requirements have increased as a result of
the development of advancing and new technologies in various sectors of human operations.
Major factors of operation which symbolize air quality are humidity, cleanness, temperature and
pressure. Conditions are selected on settings to support each process of technology. Thus general
ventilation systems cannot be used in manufacturing a biotechnology drug in a cleanroom if
maintenance of these parameters is expected to be permanent. Systematic techniques are needed
to develop unique engineering designs. Cleanroom requires complex tools of technology that
support set-up air quality parameters. The context will focus on Remicade. It is a biotechnology
drug utilized to treat particular types of arthritis such as spine arthiritis, rheumatoid arthritis and
psoriatic arthritis, along with other diseases of bowel such as ulcerative colitis and Crohn’s
disease as well as skin disease (Vasilyev et al., 2014, p. 19). The paper will evaluate the
1 Introduction
Presently cleanrooms are utilized in several industries such as biotechnology, pharmaceuticals,
defense industry, microelectronics and nanotechnology. Cleanrooms may vary from small to
large multilevel structures with big serviced utilities and equipment. Cleanroom is a managed
placement where various goods are produced and airborne particles concentration is managed to
particularized limits. As such, the killing process of contaminants of ultrafines need to be
controlled. Contaminations are created by processes, equipment, people and facilities. They must
be constantly eliminated from the air. The air cleanliness level in a room should be controlled by
standards. ISO 14644 is the most regularly applied standard (Brahm, 2012).
The primary cleanroom function is to safeguard the produced product from contamination. In
manufacturing of a biotechnology drug, manufacturer economic survival relies on the protection
of the manufactured product. Therefore, a potential contamination source should be identified,
including the working environment. Supply air quality requirements have increased as a result of
the development of advancing and new technologies in various sectors of human operations.
Major factors of operation which symbolize air quality are humidity, cleanness, temperature and
pressure. Conditions are selected on settings to support each process of technology. Thus general
ventilation systems cannot be used in manufacturing a biotechnology drug in a cleanroom if
maintenance of these parameters is expected to be permanent. Systematic techniques are needed
to develop unique engineering designs. Cleanroom requires complex tools of technology that
support set-up air quality parameters. The context will focus on Remicade. It is a biotechnology
drug utilized to treat particular types of arthritis such as spine arthiritis, rheumatoid arthritis and
psoriatic arthritis, along with other diseases of bowel such as ulcerative colitis and Crohn’s
disease as well as skin disease (Vasilyev et al., 2014, p. 19). The paper will evaluate the
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

CLEANROOM VALIDATION 4
cleanroom design, steps for validating cleanrooms, and the process required in maintaining the
validated state of the cleanroom.
2 Designing Cleanrooms
The main function of a cleanroom is to protect the product from being contaminated. In
manufacturing Remicade, the manufacturer reputation and the patients’ lives relies on the
product purity. It is thus essential to determine the potential contamination source. They may
include the raw materials, manufacturing personnel, working facilities and process equipment.
As such, it should be acknowledged that management of the operating process in the
environment, means that process equipment, raw materials and production personnel, is a setting
of successful operation of the cleanroom (Ginty et al., 2014, p. 44). Cleanroom is an environment
where the air distribution, construction materials, air supply, air supply filtration and operating
processes are controlled to regulate concentration of airborne particles to meet proper levels of
cleanliness. The department of design needs to address most challenging issues: optimum cost
against minimum risk. It refers to the cleanliness level needed to reduce risk contaminating the
product while concurrently developing a facility which operates without adding constraints to the
unit cost of the product and is economical to design.
2.1 Strategy and Purpose of design
The following are the purposes of a pharmaceutical cleanroom design in a production facility:
Eliminating the environment external to cleanrooms suite
Dilution or removal of contamination emerging from the process of manufacturing
Dilution or removal of contamination emerging from production personnel
Controlling hazards emerging from the product
Containment of cross-contamination of product-to-product
cleanroom design, steps for validating cleanrooms, and the process required in maintaining the
validated state of the cleanroom.
2 Designing Cleanrooms
The main function of a cleanroom is to protect the product from being contaminated. In
manufacturing Remicade, the manufacturer reputation and the patients’ lives relies on the
product purity. It is thus essential to determine the potential contamination source. They may
include the raw materials, manufacturing personnel, working facilities and process equipment.
As such, it should be acknowledged that management of the operating process in the
environment, means that process equipment, raw materials and production personnel, is a setting
of successful operation of the cleanroom (Ginty et al., 2014, p. 44). Cleanroom is an environment
where the air distribution, construction materials, air supply, air supply filtration and operating
processes are controlled to regulate concentration of airborne particles to meet proper levels of
cleanliness. The department of design needs to address most challenging issues: optimum cost
against minimum risk. It refers to the cleanliness level needed to reduce risk contaminating the
product while concurrently developing a facility which operates without adding constraints to the
unit cost of the product and is economical to design.
2.1 Strategy and Purpose of design
The following are the purposes of a pharmaceutical cleanroom design in a production facility:
Eliminating the environment external to cleanrooms suite
Dilution or removal of contamination emerging from the process of manufacturing
Dilution or removal of contamination emerging from production personnel
Controlling hazards emerging from the product
Containment of cross-contamination of product-to-product
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

CLEANROOM VALIDATION 5
Personnel protection
Management and control of the flow of material via the procedure processes through
configuration and layout
Management and control of the movement of personnel by improving the connection and
arrangement of each rooms
General operation safety by managing egress and entry of materials and personnel
Establishing best comfort conditions for employees
Developing unique products environmental conditions
Adaptation of process equipment and plant to guarantee easy utilization, safety and good
maintenance access
Efficient control of room conditions
All of the above purposes are essential, and cleanrooms should be structured to function and
overlap well. Thus, it is essential to examine all requirements and establish the solution in an
organized way (Fernández-Pérez et al., 2017). A simple step plan can be outlined as follows:
Evaluate the stages of production
Develop flow diagrams of processes
Define operations connected to the rooms
Define requirements of environmental quality
Measure process, production and space requirements
Develop association diagrams of the room
Define the needs of accommodation
Create schemes and layouts
Develop specifications and designs
Personnel protection
Management and control of the flow of material via the procedure processes through
configuration and layout
Management and control of the movement of personnel by improving the connection and
arrangement of each rooms
General operation safety by managing egress and entry of materials and personnel
Establishing best comfort conditions for employees
Developing unique products environmental conditions
Adaptation of process equipment and plant to guarantee easy utilization, safety and good
maintenance access
Efficient control of room conditions
All of the above purposes are essential, and cleanrooms should be structured to function and
overlap well. Thus, it is essential to examine all requirements and establish the solution in an
organized way (Fernández-Pérez et al., 2017). A simple step plan can be outlined as follows:
Evaluate the stages of production
Develop flow diagrams of processes
Define operations connected to the rooms
Define requirements of environmental quality
Measure process, production and space requirements
Develop association diagrams of the room
Define the needs of accommodation
Create schemes and layouts
Develop specifications and designs

CLEANROOM VALIDATION 6
Carry out a comprehensive construction and design process.
The above steps can be conducted at various detail levels due to the scale, physical size and
complexity of a facility. It is essential to define roles of all the involved parts (Fischer et al.,
2013, p. 5475).
2.2 Cleanroom Layout
Cleanrooms are constructed as suite of rooms which have defined purposes in
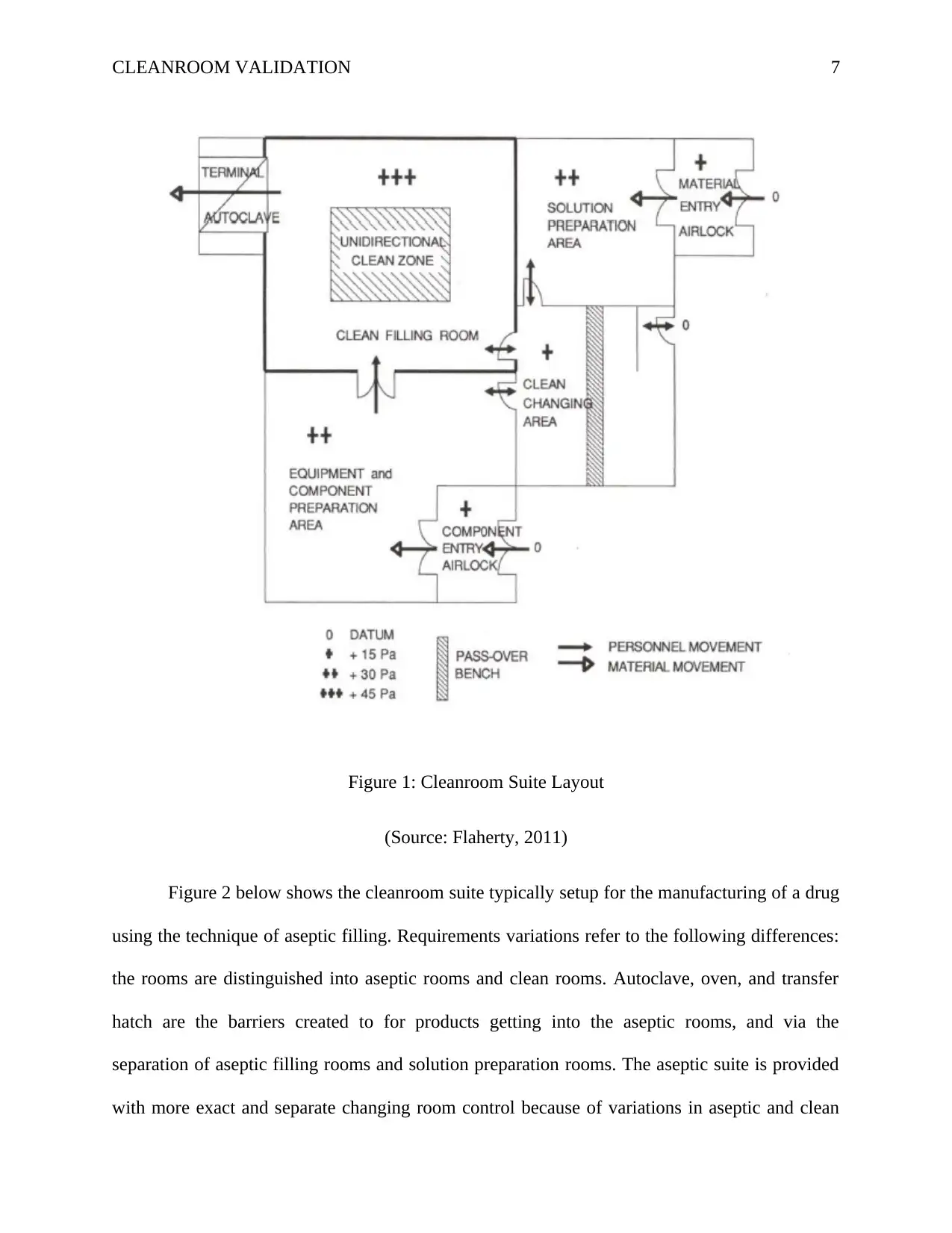
pharmaceutical manufacturing. The figures 1 and 2 illustrates this phenomenon. Figure 1 shows
a designed of cleanroom suite that meets the requirements manufacturing the drug that can be
sterilized terminally. Any worker should go through the clean changing area before entering the
cleanroom suite. In the clean changing area, the worker changes the clothes and wear the
cleanroom attire. Components and other raw materials are brought in through their respective
entry airlocks. Standard procedures are employed in the airlocks to reduce contamination from
outside. Preparation of solutions is done in the ‘Solution Preparation’ room and then indirectly or
directly transferred via mobile containers or pipes to the ‘Clean Filling’ room for filling
procedures to be undertaken. The closures and containers are cleaned and prepared in the
‘component preparation’ room and transferred using conveyor system or manually to the filling
stage. In the ‘Clean Filling’ room, which is unidirectional flow clean zone, containers are filled
and packed and transferred via the autoclave terminal sterilization.
Carry out a comprehensive construction and design process.
The above steps can be conducted at various detail levels due to the scale, physical size and
complexity of a facility. It is essential to define roles of all the involved parts (Fischer et al.,
2013, p. 5475).
2.2 Cleanroom Layout
Cleanrooms are constructed as suite of rooms which have defined purposes in
pharmaceutical manufacturing. The figures 1 and 2 illustrates this phenomenon. Figure 1 shows
a designed of cleanroom suite that meets the requirements manufacturing the drug that can be
sterilized terminally. Any worker should go through the clean changing area before entering the
cleanroom suite. In the clean changing area, the worker changes the clothes and wear the
cleanroom attire. Components and other raw materials are brought in through their respective
entry airlocks. Standard procedures are employed in the airlocks to reduce contamination from
outside. Preparation of solutions is done in the ‘Solution Preparation’ room and then indirectly or
directly transferred via mobile containers or pipes to the ‘Clean Filling’ room for filling
procedures to be undertaken. The closures and containers are cleaned and prepared in the
‘component preparation’ room and transferred using conveyor system or manually to the filling
stage. In the ‘Clean Filling’ room, which is unidirectional flow clean zone, containers are filled
and packed and transferred via the autoclave terminal sterilization.
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide
CLEANROOM VALIDATION 7
Figure 1: Cleanroom Suite Layout
(Source: Flaherty, 2011)
Figure 2 below shows the cleanroom suite typically setup for the manufacturing of a drug
using the technique of aseptic filling. Requirements variations refer to the following differences:
the rooms are distinguished into aseptic rooms and clean rooms. Autoclave, oven, and transfer
hatch are the barriers created to for products getting into the aseptic rooms, and via the
separation of aseptic filling rooms and solution preparation rooms. The aseptic suite is provided
with more exact and separate changing room control because of variations in aseptic and clean
Figure 1: Cleanroom Suite Layout
(Source: Flaherty, 2011)
Figure 2 below shows the cleanroom suite typically setup for the manufacturing of a drug
using the technique of aseptic filling. Requirements variations refer to the following differences:
the rooms are distinguished into aseptic rooms and clean rooms. Autoclave, oven, and transfer
hatch are the barriers created to for products getting into the aseptic rooms, and via the
separation of aseptic filling rooms and solution preparation rooms. The aseptic suite is provided
with more exact and separate changing room control because of variations in aseptic and clean
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

CLEANROOM VALIDATION 8
suites environmental control. Unidirectional flow workstation can be used as a substitute of
isolator.
Figure 2: Aseptic Filling Room Suites
(Source: Flaherty, 2011)
Method, quantity, and quality of air in the room, amount if incoming contamination from
neighboring rooms, and the amount if contamination released in the rooms makes it difficult to
achieve the correct internal environment cleanliness level.
suites environmental control. Unidirectional flow workstation can be used as a substitute of
isolator.
Figure 2: Aseptic Filling Room Suites
(Source: Flaherty, 2011)
Method, quantity, and quality of air in the room, amount if incoming contamination from
neighboring rooms, and the amount if contamination released in the rooms makes it difficult to
achieve the correct internal environment cleanliness level.

CLEANROOM VALIDATION 9
2.3 Changing room
In the current cleanroom, the management is required to define what employees should do. The
discipline in operators and workers by which the full contamination management operation is
maximized should be followed strictly (Kumar, Singh and Banerjee, 2015, p. 201). Influence of
employees begins in the inconstant zones when movements should increase from black via grey
to white areas. Employees change and keep outer attire in the black area. These rooms are
usually locked. Storage space should be created to store heavy and wet outdoor footwear and
clothing. Cleaning of the floor should be done quickly and regularly. Entryways should be
secured by control of contamination. The black area change room may be situated far from
cleanrooms. They should be located near the staff entryway to the building. When applicable, the
experts can stay at the grey zone. Separate female and male areas are required if personal private
clothing is transformed for uniform underclothes (Vasiliev et al., 2013). Where a complete cloth
change is effected, a personal protected locker will be needed. The flow continues to proper
utilization and storage of cleanroom clothing, where the set up will rely om the choice of
clothing and change regularity.
The grey zone should be made available for employees to remove make-up and scrub-up. The
method of administering cleanroom attires may vary. It may be simple bars with hangers or
vertical cabinets. Floor covering should be controlled from contamination. The white zone is
room used by employees to change into their cleanroom shoe. Generally, the whole changing
environment involves flow of air moving from white area where there is clean air via grey to
black area (Kurniati, Yeh and Lin, 2015, p. 248). Besides, the changing room should consist of
facilities for administering cleaned mask, facilities for storing unclean items, cleaned sterile attire
2.3 Changing room
In the current cleanroom, the management is required to define what employees should do. The
discipline in operators and workers by which the full contamination management operation is
maximized should be followed strictly (Kumar, Singh and Banerjee, 2015, p. 201). Influence of
employees begins in the inconstant zones when movements should increase from black via grey
to white areas. Employees change and keep outer attire in the black area. These rooms are
usually locked. Storage space should be created to store heavy and wet outdoor footwear and
clothing. Cleaning of the floor should be done quickly and regularly. Entryways should be
secured by control of contamination. The black area change room may be situated far from
cleanrooms. They should be located near the staff entryway to the building. When applicable, the
experts can stay at the grey zone. Separate female and male areas are required if personal private
clothing is transformed for uniform underclothes (Vasiliev et al., 2013). Where a complete cloth
change is effected, a personal protected locker will be needed. The flow continues to proper
utilization and storage of cleanroom clothing, where the set up will rely om the choice of
clothing and change regularity.
The grey zone should be made available for employees to remove make-up and scrub-up. The
method of administering cleanroom attires may vary. It may be simple bars with hangers or
vertical cabinets. Floor covering should be controlled from contamination. The white zone is
room used by employees to change into their cleanroom shoe. Generally, the whole changing
environment involves flow of air moving from white area where there is clean air via grey to
black area (Kurniati, Yeh and Lin, 2015, p. 248). Besides, the changing room should consist of
facilities for administering cleaned mask, facilities for storing unclean items, cleaned sterile attire
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

CLEANROOM VALIDATION 10
and gloves. Showers may be offered for routine or emergency use. They are located in zones
where there are individual contamination risks by harmful elements.
2.4 Material distribution technology
Movement of materials throughout the process of pharmaceutical technology is essential, but
their transfer and entrance in the cleanrooms must be managed to avoid contaminations. Raw
materials should be packed and produced in clean environment. Where paper is required, it
should be eliminated and the details purified well before allowing material to enter the
cleanroom. Airlocks should be used to transfer materials, and if possible, special trolleys or
vehicles should be utilized (Tidstam et al., 2015, p. 321). Plastic pallets should be used when
necessary. Smaller size materials should be moved via air lock hatches, using unique trays. It is
important to present different carriers with dead plate transfers at the entry point of the
cleanroom to prevent carrying contamination from grey to white zone. In addition, it is necessary
to filter fluid materials at the use point to support particles absence when in the production
process.
It should be acknowledged that particular powder based methods, for instance vial filling and
tableting are utilized when managing unrefined material. Preventing the entry of unknown
unrefined elements can be the remedy in these circumstances. It is essential to minimize the risks
to the environment and staffs (Merkulov et al., 2014, p. 19). The following content investigates
the main construction points against the draft of the cleanroom description discussed above.
2.4.1 Air supply
Air distribution, air supply and filtration are connected. Cleanliness decisions affect the
requirements of filtration which in turn affects quantities of air supply. It should be recognized
and gloves. Showers may be offered for routine or emergency use. They are located in zones
where there are individual contamination risks by harmful elements.
2.4 Material distribution technology
Movement of materials throughout the process of pharmaceutical technology is essential, but
their transfer and entrance in the cleanrooms must be managed to avoid contaminations. Raw
materials should be packed and produced in clean environment. Where paper is required, it
should be eliminated and the details purified well before allowing material to enter the
cleanroom. Airlocks should be used to transfer materials, and if possible, special trolleys or
vehicles should be utilized (Tidstam et al., 2015, p. 321). Plastic pallets should be used when
necessary. Smaller size materials should be moved via air lock hatches, using unique trays. It is
important to present different carriers with dead plate transfers at the entry point of the
cleanroom to prevent carrying contamination from grey to white zone. In addition, it is necessary
to filter fluid materials at the use point to support particles absence when in the production
process.
It should be acknowledged that particular powder based methods, for instance vial filling and
tableting are utilized when managing unrefined material. Preventing the entry of unknown
unrefined elements can be the remedy in these circumstances. It is essential to minimize the risks
to the environment and staffs (Merkulov et al., 2014, p. 19). The following content investigates
the main construction points against the draft of the cleanroom description discussed above.
2.4.1 Air supply
Air distribution, air supply and filtration are connected. Cleanliness decisions affect the
requirements of filtration which in turn affects quantities of air supply. It should be recognized
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

CLEANROOM VALIDATION 11
that making decisions on a single aspect will affect the others (Sukhanov and Petruchuk, 2018, p.
164). The number of resources such as steam, power and frozen water, and the size of air
managing plant affects the volume of air in every zone. Besides, they will determine the services
areas’ size needed to accommodate plant rooms and ductwork to accommodate production tools.
At the start of the Heating-Ventilation-Air Conditioner (HVAC) design, the air quantity to be
supplied to a room should be decided. Engineers educated to compute their designs with
correctness are normally disappointed due to unspecific cleanroom design character. The
validation needs to meet required class limits are adequate but no equation to link change rates of
air to cleanliness level (Stoller et al., 2018, p. 2335). The designer should assist with skills that
gather exact information about the operations character in the room, the tools nature, number of
participating individuals, and the room condition where it is to be examined. Then the needed air
quantity should be decided. Utilization of overpressure is a substantial factor in the prevention of
unrefined accumulation in cleanroom. Pressure gradients in room suites with varying levels of
cleanliness can be designed. Normally difference of pressure between neighboring rooms is
around 12 Pa. Big overpressures can cause difficulties in opening doors and noise in the gaps.
The pressure difference between neighboring rooms should be maintained at 5 Pa.
2.4.2 Air distribution
In the cleanroom, there are two types of air distribution. Turbulent flow is the first type.
that making decisions on a single aspect will affect the others (Sukhanov and Petruchuk, 2018, p.
164). The number of resources such as steam, power and frozen water, and the size of air
managing plant affects the volume of air in every zone. Besides, they will determine the services
areas’ size needed to accommodate plant rooms and ductwork to accommodate production tools.
At the start of the Heating-Ventilation-Air Conditioner (HVAC) design, the air quantity to be
supplied to a room should be decided. Engineers educated to compute their designs with
correctness are normally disappointed due to unspecific cleanroom design character. The
validation needs to meet required class limits are adequate but no equation to link change rates of
air to cleanliness level (Stoller et al., 2018, p. 2335). The designer should assist with skills that
gather exact information about the operations character in the room, the tools nature, number of
participating individuals, and the room condition where it is to be examined. Then the needed air
quantity should be decided. Utilization of overpressure is a substantial factor in the prevention of
unrefined accumulation in cleanroom. Pressure gradients in room suites with varying levels of
cleanliness can be designed. Normally difference of pressure between neighboring rooms is
around 12 Pa. Big overpressures can cause difficulties in opening doors and noise in the gaps.
The pressure difference between neighboring rooms should be maintained at 5 Pa.
2.4.2 Air distribution
In the cleanroom, there are two types of air distribution. Turbulent flow is the first type.

CLEANROOM VALIDATION 12
Figure3. Turbulent flow air distribution
(Source: Mohammadi et al., 2015, p. 304)
It is applied in cleanrooms where outlets of terminal make only a part of the total ceiling zone
located to fit the general room transfer or single process requirement. Extracts may be placed at
low wall levels or at the ceiling. Flows of air are foreseeable but challenging to assure. Levels of
cleanliness such as class 1000 can be accomplished when high-efficiency particle stopper
(HEPA) filters are set up in terminal housings. Laminar flow (unidirectional down flow) is the
second type. It is needed for facilities which require better conditions and class 100. Testing is a
necessity when laminar flow is being used. With a vertical flow of air containing a speed of 0.45
m/s, transfer of particle is easier to foretell.
Figure3. Turbulent flow air distribution
(Source: Mohammadi et al., 2015, p. 304)
It is applied in cleanrooms where outlets of terminal make only a part of the total ceiling zone
located to fit the general room transfer or single process requirement. Extracts may be placed at
low wall levels or at the ceiling. Flows of air are foreseeable but challenging to assure. Levels of
cleanliness such as class 1000 can be accomplished when high-efficiency particle stopper
(HEPA) filters are set up in terminal housings. Laminar flow (unidirectional down flow) is the
second type. It is needed for facilities which require better conditions and class 100. Testing is a
necessity when laminar flow is being used. With a vertical flow of air containing a speed of 0.45
m/s, transfer of particle is easier to foretell.
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide
1 out of 34
Your All-in-One AI-Powered Toolkit for Academic Success.
+13062052269
info@desklib.com
Available 24*7 on WhatsApp / Email
![[object Object]](/_next/static/media/star-bottom.7253800d.svg)
Unlock your academic potential
Copyright © 2020–2026 A2Z Services. All Rights Reserved. Developed and managed by ZUCOL.