Development, Validation, and HPLC Analysis of Ethinyl Estradiol (EE)
VerifiedAdded on 2023/06/15
|38
|10506
|131
Report
AI Summary
This report presents the development and validation of a simple, rapid, economical, and reliable high-performance liquid chromatographic (HPLC) method for the quantitation of alpha ethinyl estradiol (EE) in both coated and non-coated tablets. The method was validated according to ICH guidelines, optimizing a mobile phase of 60% acetonitrile and 40% water with a flow rate of 1.25 ml/min and UV detection at 294 nm, resulting in a retention time of 3.71 min for EE. The method demonstrated linearity with a correlation coefficient of 0.9872 and was applied to estimate EE content in different brands like Rigevidon, Microgynon, and Cilest, revealing varying EE concentrations. Further analysis, including recovery studies with varying EE concentrations, confirmed the method's accuracy and robustness. The developed HPLC method offers advantages over existing methods and is suitable for quality control assays of EE in tablet formulations, exhibiting satisfactory validation results in terms of linearity, accuracy, precision, and robustness.

Method development, validation and analysis of
ethinyl estrdiol by HPLC
1
ethinyl estrdiol by HPLC
1
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

Table of contents:
Sr. No. Contents Page no.
1 Introduction 1
1.1 Introduction to Ethynyl estradiol 1
1.1.1 Generic names 1
1.1.2 Brand names 1
1.1.3 Chemistry 2
1.2 Validation of analytical procedures 3
1.2.1 Specificity 3
1.2.2 Linearity 3
1.2.3 Range 4
1.2.4 Accuracy 4
1.2.5 Precision 4
1.2.6 Detection limit 5
1.2.7 Quantitation limit 5
1.2.8 Robustness 5
1.2.9 System suitability testing 6
1.3 HPLC 6
1.3.1 Instrumentation 8
1.4 Literature review 9
2 Aims and strategy 11
3 Methodology 14
3.1 Instruments and apparatus 14
3.2 Reagents and materials 14
3.3 Preparations and solutions 14
3.3.1 Preparation of stock solution of EE 14
3.3.2 Preparation of sample solution 15
3.4 Chromatographic conditions 15
3.5 Detection of wavelength 15
3.6 Method development and validation 15
3.6.1 Optimization of parameters 15
3.6.2 Preparation of calibration curve 15
3.6.3 Accuracy 16
3.6.4 Limit of detection (LOD) and limit of quantification
(LOQ)
16
3.6.5 Robustness 16
3.7 Assay 16
4 Results and discussion 18
4.1 Method development 18
4.2 Method validation 21
4.2.1 Linearity and range 21
4.2.2 Accuracy 25
4.2.3 Limit of detection & Limit of quantitation (LOD & LOQ) 26
4.3 Assay 26
5 Summary and conclusion 32
6 References 33
2
Sr. No. Contents Page no.
1 Introduction 1
1.1 Introduction to Ethynyl estradiol 1
1.1.1 Generic names 1
1.1.2 Brand names 1
1.1.3 Chemistry 2
1.2 Validation of analytical procedures 3
1.2.1 Specificity 3
1.2.2 Linearity 3
1.2.3 Range 4
1.2.4 Accuracy 4
1.2.5 Precision 4
1.2.6 Detection limit 5
1.2.7 Quantitation limit 5
1.2.8 Robustness 5
1.2.9 System suitability testing 6
1.3 HPLC 6
1.3.1 Instrumentation 8
1.4 Literature review 9
2 Aims and strategy 11
3 Methodology 14
3.1 Instruments and apparatus 14
3.2 Reagents and materials 14
3.3 Preparations and solutions 14
3.3.1 Preparation of stock solution of EE 14
3.3.2 Preparation of sample solution 15
3.4 Chromatographic conditions 15
3.5 Detection of wavelength 15
3.6 Method development and validation 15
3.6.1 Optimization of parameters 15
3.6.2 Preparation of calibration curve 15
3.6.3 Accuracy 16
3.6.4 Limit of detection (LOD) and limit of quantification
(LOQ)
16
3.6.5 Robustness 16
3.7 Assay 16
4 Results and discussion 18
4.1 Method development 18
4.2 Method validation 21
4.2.1 Linearity and range 21
4.2.2 Accuracy 25
4.2.3 Limit of detection & Limit of quantitation (LOD & LOQ) 26
4.3 Assay 26
5 Summary and conclusion 32
6 References 33
2
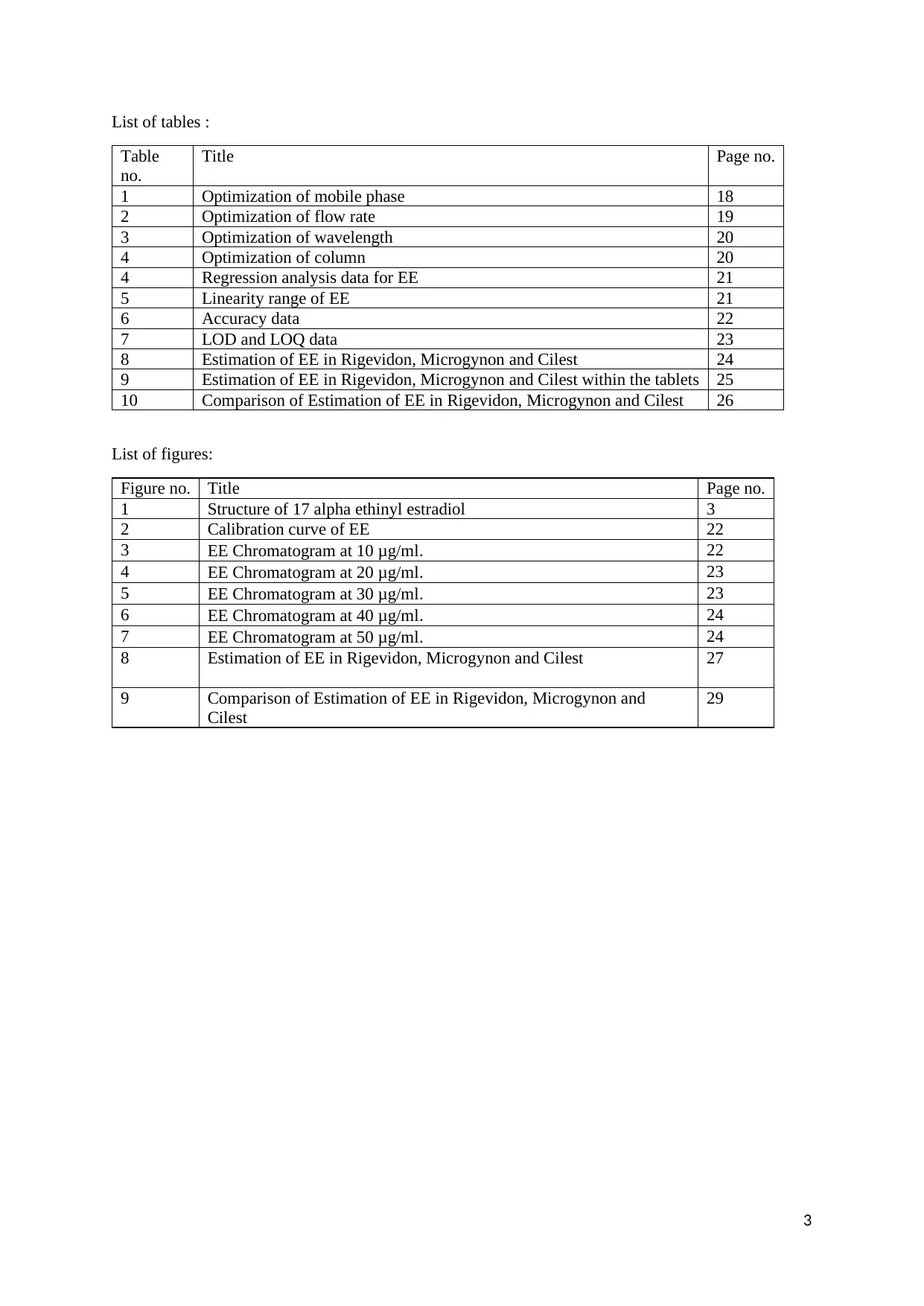
List of tables :
Table
no.
Title Page no.
1 Optimization of mobile phase 18
2 Optimization of flow rate 19
3 Optimization of wavelength 20
4 Optimization of column 20
4 Regression analysis data for EE 21
5 Linearity range of EE 21
6 Accuracy data 22
7 LOD and LOQ data 23
8 Estimation of EE in Rigevidon, Microgynon and Cilest 24
9 Estimation of EE in Rigevidon, Microgynon and Cilest within the tablets 25
10 Comparison of Estimation of EE in Rigevidon, Microgynon and Cilest 26
List of figures:
Figure no. Title Page no.
1 Structure of 17 alpha ethinyl estradiol 3
2 Calibration curve of EE 22
3 EE Chromatogram at 10 μg/ml. 22
4 EE Chromatogram at 20 μg/ml. 23
5 EE Chromatogram at 30 μg/ml. 23
6 EE Chromatogram at 40 μg/ml. 24
7 EE Chromatogram at 50 μg/ml. 24
8 Estimation of EE in Rigevidon, Microgynon and Cilest 27
9 Comparison of Estimation of EE in Rigevidon, Microgynon and
Cilest
29
3
Table
no.
Title Page no.
1 Optimization of mobile phase 18
2 Optimization of flow rate 19
3 Optimization of wavelength 20
4 Optimization of column 20
4 Regression analysis data for EE 21
5 Linearity range of EE 21
6 Accuracy data 22
7 LOD and LOQ data 23
8 Estimation of EE in Rigevidon, Microgynon and Cilest 24
9 Estimation of EE in Rigevidon, Microgynon and Cilest within the tablets 25
10 Comparison of Estimation of EE in Rigevidon, Microgynon and Cilest 26
List of figures:
Figure no. Title Page no.
1 Structure of 17 alpha ethinyl estradiol 3
2 Calibration curve of EE 22
3 EE Chromatogram at 10 μg/ml. 22
4 EE Chromatogram at 20 μg/ml. 23
5 EE Chromatogram at 30 μg/ml. 23
6 EE Chromatogram at 40 μg/ml. 24
7 EE Chromatogram at 50 μg/ml. 24
8 Estimation of EE in Rigevidon, Microgynon and Cilest 27
9 Comparison of Estimation of EE in Rigevidon, Microgynon and
Cilest
29
3
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

Abstract:
A simple, rapid, economical and reliable high-performance liquid chromatographic method has been
developed and successfully applied in quantitation of alpha ethinyl estradiol (EE) in both coated and
non-coated tablets. A HPLC method was developed and validated for the quantification of the EE in
the contraceptive pills. This method was validated as per the ICH guidelines. In this method, 60 %
Acetonitrile 40 % Water was optimized as the mobile phase and 1.25 ml/min was selected a the
mobile flow rate. UV detection was performed at 294 nm. In this method, retention time of EE was
found to be 3.71 min. Linearity of the method was found to be 0.9872. Developed method was applied
for the estimation of EE with different brands like Rigevidon, Microgynon and Cilest. Amount of EE
in Rigevidon, Microgynon and Cilest was found to be 2.90, 2.77 and 3.16 μg respectively.
Quantitation of EE in Rigevidon, Microgynon and Cilest tablets was repeated using another tablet
from the same pack. In this comparison, amount of EE in Rigevidon, Microgynon and Cilest tablets
was found to be 2.91, 4.03 and 3.44 μg respectively. Samples and standards were added in the same
amount of samples and standards to estimate amount of EE. Eight different concentrations of EE and
Rigevidon, Microgynon and Cilest tablets were used for the analysis of EE using addition between the
same samples. Good recovery was obtained at higher concentrations of EE, however less recovery
was obtained at lower concentrations of EE. The proposed HPLC method exhibited advantages over
methods available and is appropriate for quality control assays of EE in tablet formulations. The
developed HPLC method was validated in terms of linearity, accuracy, precision and robustness.
Validation reports were found to be satisfactory for the developed method.
4
A simple, rapid, economical and reliable high-performance liquid chromatographic method has been
developed and successfully applied in quantitation of alpha ethinyl estradiol (EE) in both coated and
non-coated tablets. A HPLC method was developed and validated for the quantification of the EE in
the contraceptive pills. This method was validated as per the ICH guidelines. In this method, 60 %
Acetonitrile 40 % Water was optimized as the mobile phase and 1.25 ml/min was selected a the
mobile flow rate. UV detection was performed at 294 nm. In this method, retention time of EE was
found to be 3.71 min. Linearity of the method was found to be 0.9872. Developed method was applied
for the estimation of EE with different brands like Rigevidon, Microgynon and Cilest. Amount of EE
in Rigevidon, Microgynon and Cilest was found to be 2.90, 2.77 and 3.16 μg respectively.
Quantitation of EE in Rigevidon, Microgynon and Cilest tablets was repeated using another tablet
from the same pack. In this comparison, amount of EE in Rigevidon, Microgynon and Cilest tablets
was found to be 2.91, 4.03 and 3.44 μg respectively. Samples and standards were added in the same
amount of samples and standards to estimate amount of EE. Eight different concentrations of EE and
Rigevidon, Microgynon and Cilest tablets were used for the analysis of EE using addition between the
same samples. Good recovery was obtained at higher concentrations of EE, however less recovery
was obtained at lower concentrations of EE. The proposed HPLC method exhibited advantages over
methods available and is appropriate for quality control assays of EE in tablet formulations. The
developed HPLC method was validated in terms of linearity, accuracy, precision and robustness.
Validation reports were found to be satisfactory for the developed method.
4
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

1. Introduction
1.1 Introduction to Ethynyl estradiol:
17 Alpha ethynyl estradiol (EE) is chemically described as 19-Nor-17α-pregna-1,3,5(10)-trien-20-
yne-3,17-diol. It is a derivative of 17β-estradiol. It is first orally bioactive semisynthetic estrogen
incorporated in many combined oral contraceptive pills. It is useful in the treatment and management
of menopausal symptoms and female hypogonadism (Praveen et al., 2013). However, this was not the
longer case. Lately, it was incorporated in combination formulation. It is an estrogen and it acts as
estrogen receptor agonist. Eventhough, it is a synthetic derivative of estrogen, its properties are
different from estradiol in varied ways. EE is with improved pharmacokinetic properties as compared
to the natural estradiol. EE exhibits improved bioavailability when it is orally administered.
Moreover, it is more resistant to metabolic pathways and exhibits more effects on specific parts of the
body like liver and uterus. These improved pharmacokinetic properties augmented use of this drug in
birth control pills in comparison to the estradiol (Panay et al., 2013; Unger, 2015; Kuhl, 2005)
1.1.1 Generic names:
Ethinylestradiol is the English generic name of EE. It can also be spelled as ethynylestradiol,
ethynyloestradiol, and ethinyloestradiol. All these names can be pronounced in the same fashion
(Morton and Judith, 2012; Elks, 2014).
1.1.2 Brand names:
Initially EE was marketed as individual drug with brand names Esteed, Estinyl, Feminone, Lynoral,
Menolyn, Novestrol, Palonyl, Spanestrin, and Ylestrol. However, all these formulations were
withdrawn from the market. Later, in large number EE was marketed in combination with progestins.
These combinations are being used as oral contraceptives. EE is also present in combination with
norelgestromin with brand name Ortho Evra and Xulane which are present as contraceptive patch. It
is also present in combination with etonogestrel with brand name NuvaRing which are available as
contraceptive vaginal ring.
1
1.1 Introduction to Ethynyl estradiol:
17 Alpha ethynyl estradiol (EE) is chemically described as 19-Nor-17α-pregna-1,3,5(10)-trien-20-
yne-3,17-diol. It is a derivative of 17β-estradiol. It is first orally bioactive semisynthetic estrogen
incorporated in many combined oral contraceptive pills. It is useful in the treatment and management
of menopausal symptoms and female hypogonadism (Praveen et al., 2013). However, this was not the
longer case. Lately, it was incorporated in combination formulation. It is an estrogen and it acts as
estrogen receptor agonist. Eventhough, it is a synthetic derivative of estrogen, its properties are
different from estradiol in varied ways. EE is with improved pharmacokinetic properties as compared
to the natural estradiol. EE exhibits improved bioavailability when it is orally administered.
Moreover, it is more resistant to metabolic pathways and exhibits more effects on specific parts of the
body like liver and uterus. These improved pharmacokinetic properties augmented use of this drug in
birth control pills in comparison to the estradiol (Panay et al., 2013; Unger, 2015; Kuhl, 2005)
1.1.1 Generic names:
Ethinylestradiol is the English generic name of EE. It can also be spelled as ethynylestradiol,
ethynyloestradiol, and ethinyloestradiol. All these names can be pronounced in the same fashion
(Morton and Judith, 2012; Elks, 2014).
1.1.2 Brand names:
Initially EE was marketed as individual drug with brand names Esteed, Estinyl, Feminone, Lynoral,
Menolyn, Novestrol, Palonyl, Spanestrin, and Ylestrol. However, all these formulations were
withdrawn from the market. Later, in large number EE was marketed in combination with progestins.
These combinations are being used as oral contraceptives. EE is also present in combination with
norelgestromin with brand name Ortho Evra and Xulane which are present as contraceptive patch. It
is also present in combination with etonogestrel with brand name NuvaRing which are available as
contraceptive vaginal ring.
1

1.1.3 Chemistry:
Its empirical formula is C20H24O2 and molecular weight is 296.403 g/mol. EE is practically
insoluble in water, freely soluble in solvents like ethanol and diethyl ether and sparingly soluble in
chloroform. It is a synthetic estrane steroid and estradiol derivative. At C17α position, it has ethynyl
substitution. It can also be called as 7α-ethynylestradiol or as 17α-ethynylestra-1,3,5(10)-triene-3,17β-
diol. There are different prodrugs of EE are available with varied substitutions and derivatives. These
prodrugs are known as mestranol (EE 3-methyl ether), quinestrol (EE 3-cyclopentyl ether) and
ethinylestradiol sulfonate (EE 3-isopropylsulfonate) (Elks, 2014).
Figure 1 : Structure of 17 alpha ethinyl estradiol
1.2 Validation of Analytical Procedures (ICH, 2005):
Validation of the analytical procedure is required to confirm the analytical procedure applied and to
provide support for identification, and to evaluate quality and purity of the analyte. Analytical method
development and validation can have influence quality of the data.
1.2.1 Specificity:
Specificity exercise should be conducted during the validation of the analytical procedures like
identification test, impurity profiling and quantitative assay. Procedures required to perform for
specificity will be based on the objective of the analysis. It is not possible perform one analytical
procedure for specificity hence two procedures need to be performed for specificity.
1.2.2 Linearity :
2
Its empirical formula is C20H24O2 and molecular weight is 296.403 g/mol. EE is practically
insoluble in water, freely soluble in solvents like ethanol and diethyl ether and sparingly soluble in
chloroform. It is a synthetic estrane steroid and estradiol derivative. At C17α position, it has ethynyl
substitution. It can also be called as 7α-ethynylestradiol or as 17α-ethynylestra-1,3,5(10)-triene-3,17β-
diol. There are different prodrugs of EE are available with varied substitutions and derivatives. These
prodrugs are known as mestranol (EE 3-methyl ether), quinestrol (EE 3-cyclopentyl ether) and
ethinylestradiol sulfonate (EE 3-isopropylsulfonate) (Elks, 2014).
Figure 1 : Structure of 17 alpha ethinyl estradiol
1.2 Validation of Analytical Procedures (ICH, 2005):
Validation of the analytical procedure is required to confirm the analytical procedure applied and to
provide support for identification, and to evaluate quality and purity of the analyte. Analytical method
development and validation can have influence quality of the data.
1.2.1 Specificity:
Specificity exercise should be conducted during the validation of the analytical procedures like
identification test, impurity profiling and quantitative assay. Procedures required to perform for
specificity will be based on the objective of the analysis. It is not possible perform one analytical
procedure for specificity hence two procedures need to be performed for specificity.
1.2.2 Linearity :
2
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

Linearity should be demonstrated across all the analytical procedures. Linearity can be performed in
two ways. In one method, standard stock of drug substance should be diluted and, in another method,
separate weighing of the drug substance should be done. Linearity can be performed by visual
inspection of the plot of signals with respect to analyte concentration. Linear relationship should be
calculated by using suitable statistical methods by calculating regression line using least square
method. Regression line can be used as mathematical estimation of degree of linearity. The
correlation coefficient, y-intercept, slope of the regression line and residual sum of squares should be
calculated and represented. Minimum five concentrations should be used for the estimation of the
linearity.
1.2.3 Range:
Range should be estimated based on the linearity studies and planed application of the analytical
procedure. Analytical procedure should provide adequate degree of linearity, accuracy and precision
within or at the extreme range of analytical procedures.
1.2.4 Accuracy:
Accuracy should be established for the given analyte throughout the range of analytical procedures
and analyte concentrations. Accuracy can be measured by using assays on drug substances and drug
products. Accuracy can also be determined by spiking impurities with the known substances.
Accuracy should be estimated by using at least 9 estimations with minimum of 3 different
concentration levels. These concentration levels should be within specified range and 3 concentrations
should be used in replicates. Accuracy can be represented in two ways like 1) percent recovery which
estimates known added amount of substance to be analysed to the sample, 2) variance among mean
and accepted true value in addition to the confidence intervals.
1.2.5 Precision:
Precision is required for the validation of analytical tests and quantitative determination of impurities.
Precision includes repeatability, intermediate precision and reproducibility. Repeatability can be
measured either of the two methods: a) minimum 9 determinations should be preformed comprising
3
two ways. In one method, standard stock of drug substance should be diluted and, in another method,
separate weighing of the drug substance should be done. Linearity can be performed by visual
inspection of the plot of signals with respect to analyte concentration. Linear relationship should be
calculated by using suitable statistical methods by calculating regression line using least square
method. Regression line can be used as mathematical estimation of degree of linearity. The
correlation coefficient, y-intercept, slope of the regression line and residual sum of squares should be
calculated and represented. Minimum five concentrations should be used for the estimation of the
linearity.
1.2.3 Range:
Range should be estimated based on the linearity studies and planed application of the analytical
procedure. Analytical procedure should provide adequate degree of linearity, accuracy and precision
within or at the extreme range of analytical procedures.
1.2.4 Accuracy:
Accuracy should be established for the given analyte throughout the range of analytical procedures
and analyte concentrations. Accuracy can be measured by using assays on drug substances and drug
products. Accuracy can also be determined by spiking impurities with the known substances.
Accuracy should be estimated by using at least 9 estimations with minimum of 3 different
concentration levels. These concentration levels should be within specified range and 3 concentrations
should be used in replicates. Accuracy can be represented in two ways like 1) percent recovery which
estimates known added amount of substance to be analysed to the sample, 2) variance among mean
and accepted true value in addition to the confidence intervals.
1.2.5 Precision:
Precision is required for the validation of analytical tests and quantitative determination of impurities.
Precision includes repeatability, intermediate precision and reproducibility. Repeatability can be
measured either of the two methods: a) minimum 9 determinations should be preformed comprising
3
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

of 3 concentrations with three replicates each, or b) minimum three determinations with 100 %
concentrations. Use of intended precision should be based on circumstances under which analytical
procedure is being carried out. Effects of random events should be established on the analytical
procedure. Different variables like days, analysts and equipments should be established. It is not
mandatory to study these variables, however these can be studied in matrix experimental setup.
Reproducibility can be evaluated by performing inter-laboratory variables on the outcome of the
experiment. Data like standard deviation, coefficient of variation and confidence interval should be
measured for measuring precision of the method.
1.2.6 Detection limit:
Based on the application of non-instrumental or instrumental methods, detection limit can be
determined by using different methods like based on visual evaluation, based on signal-to-noise ratio
and based on the standard deviation of the response and the slope. Based on the standard deviation of
the response and the slope can be estimated by using methods based on the standard deviation of the
blank and based on the calibration curve. Detection limit value and method used for estimating
detection limit should be represented in the results. Respective chromatograms can be considered as
acceptable, if visual evaluation or signal to noise ration used as method for estimating detection limit.
1.2.7 Quantitation limit:
Based on the application of non-instrumental or instrumental methods, quantitation limit can be
determined by using different methods like based on Visual evaluation, based on signal-to-noise ratio
and based on the Standard Deviation of the Response and the Slope. Based on the Standard Deviation
of the Response and the Slope can be estimated by using methods based on the Standard Deviation of
the Blank and based on the Calibration Curve. Quantitation limit value and method used for
estimating detection limit should be represented in the results. Quantitation limit can be validated by
analysing different samples with concentration near quantitation limit.
1.2.8 Robustness:
4
concentrations. Use of intended precision should be based on circumstances under which analytical
procedure is being carried out. Effects of random events should be established on the analytical
procedure. Different variables like days, analysts and equipments should be established. It is not
mandatory to study these variables, however these can be studied in matrix experimental setup.
Reproducibility can be evaluated by performing inter-laboratory variables on the outcome of the
experiment. Data like standard deviation, coefficient of variation and confidence interval should be
measured for measuring precision of the method.
1.2.6 Detection limit:
Based on the application of non-instrumental or instrumental methods, detection limit can be
determined by using different methods like based on visual evaluation, based on signal-to-noise ratio
and based on the standard deviation of the response and the slope. Based on the standard deviation of
the response and the slope can be estimated by using methods based on the standard deviation of the
blank and based on the calibration curve. Detection limit value and method used for estimating
detection limit should be represented in the results. Respective chromatograms can be considered as
acceptable, if visual evaluation or signal to noise ration used as method for estimating detection limit.
1.2.7 Quantitation limit:
Based on the application of non-instrumental or instrumental methods, quantitation limit can be
determined by using different methods like based on Visual evaluation, based on signal-to-noise ratio
and based on the Standard Deviation of the Response and the Slope. Based on the Standard Deviation
of the Response and the Slope can be estimated by using methods based on the Standard Deviation of
the Blank and based on the Calibration Curve. Quantitation limit value and method used for
estimating detection limit should be represented in the results. Quantitation limit can be validated by
analysing different samples with concentration near quantitation limit.
1.2.8 Robustness:
4

Robustness evaluation of the analytical method should be considered during the development phase
and it should decided based on the procedure implemented in the study. It should demonstrate
reliability of the method. If in case, parameters to be analysed are susceptible to variation, then
analytical procedures should be controlled and related statement should be mentioned in the analytical
procedure. System suitability parameters can be established in the robustness evaluation. It makes
sure that validity of the analytical method can be maintained all the time. In case of HPLC, following
are the examples of variations in analytical procedure: PH of the mobile phase, mobile phase
composition, solvents and chemicals of varied lots or suppliers, temperature and flow rate.
1.2.9 System suitability testing:
System suitability testing is the integral component of the analytical method development. System
suitability testing is based on the concept that equipment, electronics, analytical operations and
analytes are the integral components of the analytical procedures and these should be evaluated as
such. System suitability parameters to be established in the particular analytical method depends on
the particular procedure being validated. System suitability parameters comprises of capacity factor,
repeatability, relative retention, resolution, tailing factor and theoretical plates.
1.3 Reverse Phase – High Performance Liquid Chromatography (RP- HPLC):
In RP-HPLC, pump needs to pass solvent with pressure through the column which comprises of filled
solid adsorbent material. Solvent should contain mixture of samples from which required analyte gets
separated. Components of the sample mixture interact with the adsorbent with different affinities
which results in the separation of required analyte as solvent flow out of the column. Characteristics
of RP-HPLC includes speed, greater sensitivity, improved resolution, re-usable columns, easy sample
recovery, handling and maintenance, instrumentation tends itself to automation and quantitation,
precise and reproducible results, calculations are done by integrator itself. Reverse-phase high-
performance liquid chromatography (RP-HPLC) performs separation mainly based on the
hydrophobicity. In RP-HPLC, solute in the mobile phases binds hydrophobically to the immobilised
5
and it should decided based on the procedure implemented in the study. It should demonstrate
reliability of the method. If in case, parameters to be analysed are susceptible to variation, then
analytical procedures should be controlled and related statement should be mentioned in the analytical
procedure. System suitability parameters can be established in the robustness evaluation. It makes
sure that validity of the analytical method can be maintained all the time. In case of HPLC, following
are the examples of variations in analytical procedure: PH of the mobile phase, mobile phase
composition, solvents and chemicals of varied lots or suppliers, temperature and flow rate.
1.2.9 System suitability testing:
System suitability testing is the integral component of the analytical method development. System
suitability testing is based on the concept that equipment, electronics, analytical operations and
analytes are the integral components of the analytical procedures and these should be evaluated as
such. System suitability parameters to be established in the particular analytical method depends on
the particular procedure being validated. System suitability parameters comprises of capacity factor,
repeatability, relative retention, resolution, tailing factor and theoretical plates.
1.3 Reverse Phase – High Performance Liquid Chromatography (RP- HPLC):
In RP-HPLC, pump needs to pass solvent with pressure through the column which comprises of filled
solid adsorbent material. Solvent should contain mixture of samples from which required analyte gets
separated. Components of the sample mixture interact with the adsorbent with different affinities
which results in the separation of required analyte as solvent flow out of the column. Characteristics
of RP-HPLC includes speed, greater sensitivity, improved resolution, re-usable columns, easy sample
recovery, handling and maintenance, instrumentation tends itself to automation and quantitation,
precise and reproducible results, calculations are done by integrator itself. Reverse-phase high-
performance liquid chromatography (RP-HPLC) performs separation mainly based on the
hydrophobicity. In RP-HPLC, solute in the mobile phases binds hydrophobically to the immobilised
5
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

hydrophobic ligands present on the stationary phase which is also called as sorbent. Solute can be put
on the sorbent in the presence of aqueous buffer and it can be eluted by using organic solvents. In RP-
HPLC, elution can be categorised in gradient and isocratic elution. In gradient elution, quantity of
organic solvent goes on increasing with time and in isocratic elution quantity of organic solvent
remains constant. Solutes can be eluted with increasing power of hydrophobicity of the solute
molecules. Most commonly used packing material for RP-HPLC is microparticulate porous silica.
This material helps in achieving higher velocity and it can be useful in rapid separation. Modification
need to be done in silica by using derivatized silane containing an n-alkyl hydrophobic ligand. This
modification leads to increasing hydrophobicity of the sorbent. Most commonly used ligand is C18.
C8 and C4 ligands can also be used in RP-HPLC. Retention time of analyte can be raised by adding
more amount water to the mobile phase because affinity of the hydrophobic analyte to the
hydrophobic stationary phase can be improved as compared to the affinity towards hydrophilic mobile
phase. On the other hand, retention time of analyte can be decreased by adding more amount of
organic solvent to the mobile phase. Chemical characteristics of the solute molecules also plays
important role in the separation. Analytes with the hydrophobic characteristics retained for the longer
duration because of its less interaction with the hydrophilic mobile phase. Analytes with hydrophobic
characteristics contain groups such as C-H, C-C and S-S. Solutes with more hydrophilic
characteristics retained less because of their more interaction with hydrophilic mobile phase. Analytes
with hydrophilic characteristics contain groups such as -OH, NH2, COO- and NH3. pH of mobile
phase is also important factor because it can alter hydrophobic characteristics of analyte. Hence,
buffering agent like sodium phosphate can be used to change pH. There are less chances of damage of
RP-HPLC column as compared to the normal phase chromatography. However, RP-HPLC aqueous
bases should not be used in the RP-HPLC columns. Aqueous acids can be used, however, it should
not expose to the columns for the longer duration. Immediately after the use RP-HPLC columns
should be flushed with suitable solvent to remove buffers. Required efficiency and amount of sample
loading decides the dimension of column to be selected for analysis. Resolution can be increased by
increasing column length. Internal diameter of the column can be selected based on the sample
capacity and detection sensitivity. Operating temperature can also influence resolution because
6
on the sorbent in the presence of aqueous buffer and it can be eluted by using organic solvents. In RP-
HPLC, elution can be categorised in gradient and isocratic elution. In gradient elution, quantity of
organic solvent goes on increasing with time and in isocratic elution quantity of organic solvent
remains constant. Solutes can be eluted with increasing power of hydrophobicity of the solute
molecules. Most commonly used packing material for RP-HPLC is microparticulate porous silica.
This material helps in achieving higher velocity and it can be useful in rapid separation. Modification
need to be done in silica by using derivatized silane containing an n-alkyl hydrophobic ligand. This
modification leads to increasing hydrophobicity of the sorbent. Most commonly used ligand is C18.
C8 and C4 ligands can also be used in RP-HPLC. Retention time of analyte can be raised by adding
more amount water to the mobile phase because affinity of the hydrophobic analyte to the
hydrophobic stationary phase can be improved as compared to the affinity towards hydrophilic mobile
phase. On the other hand, retention time of analyte can be decreased by adding more amount of
organic solvent to the mobile phase. Chemical characteristics of the solute molecules also plays
important role in the separation. Analytes with the hydrophobic characteristics retained for the longer
duration because of its less interaction with the hydrophilic mobile phase. Analytes with hydrophobic
characteristics contain groups such as C-H, C-C and S-S. Solutes with more hydrophilic
characteristics retained less because of their more interaction with hydrophilic mobile phase. Analytes
with hydrophilic characteristics contain groups such as -OH, NH2, COO- and NH3. pH of mobile
phase is also important factor because it can alter hydrophobic characteristics of analyte. Hence,
buffering agent like sodium phosphate can be used to change pH. There are less chances of damage of
RP-HPLC column as compared to the normal phase chromatography. However, RP-HPLC aqueous
bases should not be used in the RP-HPLC columns. Aqueous acids can be used, however, it should
not expose to the columns for the longer duration. Immediately after the use RP-HPLC columns
should be flushed with suitable solvent to remove buffers. Required efficiency and amount of sample
loading decides the dimension of column to be selected for analysis. Resolution can be increased by
increasing column length. Internal diameter of the column can be selected based on the sample
capacity and detection sensitivity. Operating temperature can also influence resolution because
6
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

temperature can affect viscosity of the mobile phase. Most of the regulatory agencies are making
compulsion to analyse the drug with RP-HPLC before its release in the market.
Advantages of RP-HPLC include : 1 ) High level of resolution can be attained using varied
chromatographic conditions, 2) structurally similar and distinct chemical compounds can be
efficiently separated, 3) high recovery and high productivity can be achieved, and 4 ) high level of
reproducibility can be achieved mainly due to stability of sorbent.
1.3.1 Instrumentation:
Various components of RP-HPLC include a solvent delivery system, including pump, sample
injection system, a chromatographic column, a detector, a strip chart recorder, data handling device
and microprocessor control. Solvent delivery system is used to pump mobile phase under pressure.
Mobile phase flow through the pump single or more reservoirs and with the uniform flow rate. Based
on the eluting power of mobile phase, RP-HPLC separation can be classified in to normal phase and
reverse phase. With increase in the polarity of the mobile, eluting power increase in the normal phase.
With the increase in the polarity of the mobile phase, eluting power decreases in the reversed phase
chromatography. Pumps are the main components of the solvent delivery system in the RP-HPLC.
Pump efficiency can have impact on retention time, reproducibility and detector sensitivity.
Displacement pump, Reciprocating pump and Pneumatic or constant pressure pump are the three
different types of pumps which can be used in RP-HPLC. Sample injection system can be used to
introduce sample into the injection port. Loop injection, Valve injection and On column injection are
the three types of sample injection system which can be used in the RP-HPLC. Material used in the
manufacture of chromatographic columns in the RP-HPLC are heavy glass or stainless-steel tubing.
These materials can be useful to withstand high pressure. Generally, these columns are 10-30 cm in
length and with 4-10 mm inside diameter. Stationary phase filled in these columns is usually with
particle diameter 25 μm or less. It is evident that columns with internal diameter of approximately 5
mm give good results. Column packing material used in the RP-HPLC is usually of small and rigid
particles. Main function of detectors used in the RP-HPLC is to monitor mobile phase. Universal
detectors and selective detectors are the two types of detectors can be used in the RP-HPLC.
7
compulsion to analyse the drug with RP-HPLC before its release in the market.
Advantages of RP-HPLC include : 1 ) High level of resolution can be attained using varied
chromatographic conditions, 2) structurally similar and distinct chemical compounds can be
efficiently separated, 3) high recovery and high productivity can be achieved, and 4 ) high level of
reproducibility can be achieved mainly due to stability of sorbent.
1.3.1 Instrumentation:
Various components of RP-HPLC include a solvent delivery system, including pump, sample
injection system, a chromatographic column, a detector, a strip chart recorder, data handling device
and microprocessor control. Solvent delivery system is used to pump mobile phase under pressure.
Mobile phase flow through the pump single or more reservoirs and with the uniform flow rate. Based
on the eluting power of mobile phase, RP-HPLC separation can be classified in to normal phase and
reverse phase. With increase in the polarity of the mobile, eluting power increase in the normal phase.
With the increase in the polarity of the mobile phase, eluting power decreases in the reversed phase
chromatography. Pumps are the main components of the solvent delivery system in the RP-HPLC.
Pump efficiency can have impact on retention time, reproducibility and detector sensitivity.
Displacement pump, Reciprocating pump and Pneumatic or constant pressure pump are the three
different types of pumps which can be used in RP-HPLC. Sample injection system can be used to
introduce sample into the injection port. Loop injection, Valve injection and On column injection are
the three types of sample injection system which can be used in the RP-HPLC. Material used in the
manufacture of chromatographic columns in the RP-HPLC are heavy glass or stainless-steel tubing.
These materials can be useful to withstand high pressure. Generally, these columns are 10-30 cm in
length and with 4-10 mm inside diameter. Stationary phase filled in these columns is usually with
particle diameter 25 μm or less. It is evident that columns with internal diameter of approximately 5
mm give good results. Column packing material used in the RP-HPLC is usually of small and rigid
particles. Main function of detectors used in the RP-HPLC is to monitor mobile phase. Universal
detectors and selective detectors are the two types of detectors can be used in the RP-HPLC.
7

Universal detectors are also known as the bulk property detectors and these detectors measure
physical property of the mobile phase either with solute or without solute. Refractive index, dielectric
constant or density can be measure using bulk property detectors. Selective detectors are also known
as solute property detectors and these detectors can measure physical properties of the detectors which
cannot displayed by mobile phase. UV absorbance, fluorescence or diffusion current are the examples
of the solute specific detectors. These detectors are approximately 1000 times more sensitive as
compared to the bulk property detectors and can detect even nanogram quantity of the sample. UV-
Vis absorbance detector is the most commonly used detector in the HPLC system (Mendham et al.,
2006; Skoog et al., 1980; Beckett and Stenlake, 1997).
1.4 Literature review:
EE and mestranol are the two most widely used synthetic contraceptive estrogens available. Mestranol
contains one extra methyl group attached to C-3. Mestranol should be converted to EE in the liver to
exhibit its pharmacological effect. It is evident that mestranol is 50 % less active as compared to EE.
EE is present in the dose range of 20 – 50 μg in oral dosage forms. EE is an oral contraceptive agent.
There are numerous contraceptive formulations available in the market containing EE along with
other drugs. Thorough literature survey revealed that various analytical methods are existing for the
estimation of EE in bulk drugs and combination formulations. New formulations were developed for
the EE with low dose levels to maintain balance between safety and efficacy. Due to presence of low
level of doses and requirement of long duration treatment, quality of EE containing formulations need
to be maintained. Quality of drugs and its formulations can be evaluated by applying analytical
methods including HPLC. Literature survey revealed that few analytical methods are available for the
quantitation for EE in the solid dosage forms (Strusiak et al., 1982). However, these methods are
associated with disadvantages like complex method of sample preparation and solvent system
preparation. A colorimetric method is available for the quantification of the EE in the solid dosage
forms. This method is based on the formation of azo dye due to condensation of the diazotized 5‐
chloro‐2, 4‐dinitroaniline with ethinyl estradiol (Mohamed et al., 1975). Hence, this method is having
less sensitivity as compared to the HPLC method. Most of the methods available for the quantitation
8
physical property of the mobile phase either with solute or without solute. Refractive index, dielectric
constant or density can be measure using bulk property detectors. Selective detectors are also known
as solute property detectors and these detectors can measure physical properties of the detectors which
cannot displayed by mobile phase. UV absorbance, fluorescence or diffusion current are the examples
of the solute specific detectors. These detectors are approximately 1000 times more sensitive as
compared to the bulk property detectors and can detect even nanogram quantity of the sample. UV-
Vis absorbance detector is the most commonly used detector in the HPLC system (Mendham et al.,
2006; Skoog et al., 1980; Beckett and Stenlake, 1997).
1.4 Literature review:
EE and mestranol are the two most widely used synthetic contraceptive estrogens available. Mestranol
contains one extra methyl group attached to C-3. Mestranol should be converted to EE in the liver to
exhibit its pharmacological effect. It is evident that mestranol is 50 % less active as compared to EE.
EE is present in the dose range of 20 – 50 μg in oral dosage forms. EE is an oral contraceptive agent.
There are numerous contraceptive formulations available in the market containing EE along with
other drugs. Thorough literature survey revealed that various analytical methods are existing for the
estimation of EE in bulk drugs and combination formulations. New formulations were developed for
the EE with low dose levels to maintain balance between safety and efficacy. Due to presence of low
level of doses and requirement of long duration treatment, quality of EE containing formulations need
to be maintained. Quality of drugs and its formulations can be evaluated by applying analytical
methods including HPLC. Literature survey revealed that few analytical methods are available for the
quantitation for EE in the solid dosage forms (Strusiak et al., 1982). However, these methods are
associated with disadvantages like complex method of sample preparation and solvent system
preparation. A colorimetric method is available for the quantification of the EE in the solid dosage
forms. This method is based on the formation of azo dye due to condensation of the diazotized 5‐
chloro‐2, 4‐dinitroaniline with ethinyl estradiol (Mohamed et al., 1975). Hence, this method is having
less sensitivity as compared to the HPLC method. Most of the methods available for the quantitation
8
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide
1 out of 38

Your All-in-One AI-Powered Toolkit for Academic Success.
+13062052269
info@desklib.com
Available 24*7 on WhatsApp / Email
![[object Object]](/_next/static/media/star-bottom.7253800d.svg)
Unlock your academic potential
Copyright © 2020–2026 A2Z Services. All Rights Reserved. Developed and managed by ZUCOL.