Cancer Drugs Approval Process in EU and USA
VerifiedAdded on 2023/03/30
|55
|14227
|222
AI Summary
This study explores the drug approval process for cancer drugs in the European Union and United States. It examines the differences and implications of the approval process, highlighting the complexities and challenges. The study recommends new ways to improve the process and suggests the possibility of joint research by FDA and EMA. The research aims to enhance knowledge and application for a more patient-centered and rapid drug approval process.
Contribute Materials
Your contribution can guide someone’s learning journey. Share your
documents today.

NAME: KALYAN CHALAMCHARLA
STDENT ID: C00243361
TITLE: CANCER DRUGS APPROVAL PROCESS IN EU AND USA
STDENT ID: C00243361
TITLE: CANCER DRUGS APPROVAL PROCESS IN EU AND USA
Secure Best Marks with AI Grader
Need help grading? Try our AI Grader for instant feedback on your assignments.

Contents
List of figure...............................................................................................................................2
Ethical considerations................................................................................................................3
Declaration.................................................................................................................................4
Abstract......................................................................................................................................5
Chapter 1 – Introduction............................................................................................................6
1.1 Background......................................................................................................................6
1.2 Problem statement............................................................................................................8
1.3 Aims and Objectives..........................................................................................................11
Aim.......................................................................................................................................11
Objectives.............................................................................................................................11
1.4 Structure of the Research...............................................................................................12
Chapter One ........................................................................................................................12
Chapter Two.........................................................................................................................12
Chapter Three.......................................................................................................................12
Chapter Four.........................................................................................................................12
Chapter Five.........................................................................................................................12
Chapter 2 – Literature review..................................................................................................12
Introduction..........................................................................................................................12
2.2 Review............................................................................................................................14
2.3 Summary........................................................................................................................19
1
List of figure...............................................................................................................................2
Ethical considerations................................................................................................................3
Declaration.................................................................................................................................4
Abstract......................................................................................................................................5
Chapter 1 – Introduction............................................................................................................6
1.1 Background......................................................................................................................6
1.2 Problem statement............................................................................................................8
1.3 Aims and Objectives..........................................................................................................11
Aim.......................................................................................................................................11
Objectives.............................................................................................................................11
1.4 Structure of the Research...............................................................................................12
Chapter One ........................................................................................................................12
Chapter Two.........................................................................................................................12
Chapter Three.......................................................................................................................12
Chapter Four.........................................................................................................................12
Chapter Five.........................................................................................................................12
Chapter 2 – Literature review..................................................................................................12
Introduction..........................................................................................................................12
2.2 Review............................................................................................................................14
2.3 Summary........................................................................................................................19
1

2.4 Literature gap.................................................................................................................20
Chapter 3 - Research methodology..........................................................................................21
3.1 Introduction....................................................................................................................21
3.2 Research philosophy......................................................................................................21
3.3 Research approach.........................................................................................................22
3.4 Design............................................................................................................................23
3.5 Data collection method..................................................................................................25
3.6 Data analysis..................................................................................................................25
3.7Summary.........................................................................................................................26
Chapter 4 Results and Discussion............................................................................................26
4.1Drug Approval in the United States................................................................................26
4.2Drug Approval in Europe................................................................................................29
Chapter 5 : Conclusion and recommendations.........................................................................40
REFERENCES.........................................................................................................................44
List of figure
Figure 1 : Drug Market approval process in United States......................................................27
Figure 2: New Drug Application..............................................................................................28
Figure 3: Generic Drugs Approval...........................................................................................29
Figure 4 : Mutual Recognition Procedure................................................................................31
Figure 5: Decentralised Procedure...........................................................................................32
2
Chapter 3 - Research methodology..........................................................................................21
3.1 Introduction....................................................................................................................21
3.2 Research philosophy......................................................................................................21
3.3 Research approach.........................................................................................................22
3.4 Design............................................................................................................................23
3.5 Data collection method..................................................................................................25
3.6 Data analysis..................................................................................................................25
3.7Summary.........................................................................................................................26
Chapter 4 Results and Discussion............................................................................................26
4.1Drug Approval in the United States................................................................................26
4.2Drug Approval in Europe................................................................................................29
Chapter 5 : Conclusion and recommendations.........................................................................40
REFERENCES.........................................................................................................................44
List of figure
Figure 1 : Drug Market approval process in United States......................................................27
Figure 2: New Drug Application..............................................................................................28
Figure 3: Generic Drugs Approval...........................................................................................29
Figure 4 : Mutual Recognition Procedure................................................................................31
Figure 5: Decentralised Procedure...........................................................................................32
2

Ethical considerations
To be ethical, research should have a scientific merit and should pass the judgement
of independently scientific committee and not by the researchers themselves. This assessment
shall be done with a peer review method that is employed by a funding agency. The research
methodology should be appropriate to research aim and the results. The relevant ongoing
research must be taken in account. Over few decades, greater insistence is provided by the
research funding authority and the ethics bodies. It has to be analysed by the journal editors.
A systemic review of prior research on the same team has to be undertaken before proceeding
to any further research on the topic. All the ethical considerations were addressed by the
researches included in this study and their validity and reliability were assured as well.
Declaration
3
To be ethical, research should have a scientific merit and should pass the judgement
of independently scientific committee and not by the researchers themselves. This assessment
shall be done with a peer review method that is employed by a funding agency. The research
methodology should be appropriate to research aim and the results. The relevant ongoing
research must be taken in account. Over few decades, greater insistence is provided by the
research funding authority and the ethics bodies. It has to be analysed by the journal editors.
A systemic review of prior research on the same team has to be undertaken before proceeding
to any further research on the topic. All the ethical considerations were addressed by the
researches included in this study and their validity and reliability were assured as well.
Declaration
3
Secure Best Marks with AI Grader
Need help grading? Try our AI Grader for instant feedback on your assignments.

Abstract
Drug approval process in Europe and United States is a lengthy and complicated
process that influence the expenditure, waiting time and market availability of the drugs in a
a drastic way. While quality, risk and safety issues are common with the drugs approved by
FDA of United States ad EMA of Europe – these agencies continues to evolve with the
demands for new generations of drugs. Oncology drugs, used for different cancer conditions
are novelistic and they involve a pathbreaking research approach with adherence to quality
constraints and market demands. The problems though, lies within each step of the drug
approval process from its initial development to its administration to the patients and the drug
reviews related to the same. The study aims to drug approval process in cancer drugs in
European Union and United States of America. The study uses a secondary data collection
and a mixed metod analysis to gather and scrutinize te data. This research study explores the
research problem from new perspectives and attempts to understand the cancer drug approval
process of both the agengies while highlighting the complexities and implication present with
the drug development and drug approval framework. The study recommends new ways of
dealing with the issues associated with the drug approval process in general and cancer drg
approval process in specific as it takes into count, a possibility of joint research by FDA and
EMA, in order to validate the results and decrease the research expenditure. The research
study is determined to enance knowledge and application to a more patient centred care and
more rapid but quality drug approval process.
4
Drug approval process in Europe and United States is a lengthy and complicated
process that influence the expenditure, waiting time and market availability of the drugs in a
a drastic way. While quality, risk and safety issues are common with the drugs approved by
FDA of United States ad EMA of Europe – these agencies continues to evolve with the
demands for new generations of drugs. Oncology drugs, used for different cancer conditions
are novelistic and they involve a pathbreaking research approach with adherence to quality
constraints and market demands. The problems though, lies within each step of the drug
approval process from its initial development to its administration to the patients and the drug
reviews related to the same. The study aims to drug approval process in cancer drugs in
European Union and United States of America. The study uses a secondary data collection
and a mixed metod analysis to gather and scrutinize te data. This research study explores the
research problem from new perspectives and attempts to understand the cancer drug approval
process of both the agengies while highlighting the complexities and implication present with
the drug development and drug approval framework. The study recommends new ways of
dealing with the issues associated with the drug approval process in general and cancer drg
approval process in specific as it takes into count, a possibility of joint research by FDA and
EMA, in order to validate the results and decrease the research expenditure. The research
study is determined to enance knowledge and application to a more patient centred care and
more rapid but quality drug approval process.
4

Chapter 1 – Introduction
1.1 Background
In drug developmental process, the chemists begin their research by searching the
right chemicals for drug engineering. They scrutinize the medical literatures and the libraries
to find the right mixture of choices for their new medicine. This delicate process has three
different steps: (a) development and regulation of compound libraries (Corsello et al;2017)
(b) the specific (precise) assay development (c) thorough screening of each process (Forli et
al;2016). Assays quantify the interaction of biological targets with a compound that the
scientists and pharmacists are examining. Testing for increasing biological targets that
encompass many different biochemical entities need some very refined screening methods
(Surmeier et al;2016). The researchers nowadays use robotics in order to test thousands of
different chemical compounds with binding and functional assays (Katsuno et al;2015).
Manof eachgy of the times - the academic researchers apply expert knowledge in precise
pathways that will guide the assay development in the pharmaceutics industry. The
manufacture and approval of cancer drugs is a more critical process as compared to approval
of other drugs.
For the drugs of small molecules, the approval of drugs comprise of a long and very
exhaustive journey through medicine discovery, preclinical tests, basic research (Frye et al;
2015), that progressively complicate human clinical trials and a regulatory approval (by
‘Food and Drug Administration of United States’). The drug approval administration takes
many years to approve a drug for marketing. Owing to this complexity, the drug discovery
(Pascoalino et al;2016), development and approval becomes a risky process in the scientific
industry, and it impends a major challenge to the biomedical industry. Much of the risks
leading to failures, account for almost about seventeen per cent of total research
‘development costs’.
5
1.1 Background
In drug developmental process, the chemists begin their research by searching the
right chemicals for drug engineering. They scrutinize the medical literatures and the libraries
to find the right mixture of choices for their new medicine. This delicate process has three
different steps: (a) development and regulation of compound libraries (Corsello et al;2017)
(b) the specific (precise) assay development (c) thorough screening of each process (Forli et
al;2016). Assays quantify the interaction of biological targets with a compound that the
scientists and pharmacists are examining. Testing for increasing biological targets that
encompass many different biochemical entities need some very refined screening methods
(Surmeier et al;2016). The researchers nowadays use robotics in order to test thousands of
different chemical compounds with binding and functional assays (Katsuno et al;2015).
Manof eachgy of the times - the academic researchers apply expert knowledge in precise
pathways that will guide the assay development in the pharmaceutics industry. The
manufacture and approval of cancer drugs is a more critical process as compared to approval
of other drugs.
For the drugs of small molecules, the approval of drugs comprise of a long and very
exhaustive journey through medicine discovery, preclinical tests, basic research (Frye et al;
2015), that progressively complicate human clinical trials and a regulatory approval (by
‘Food and Drug Administration of United States’). The drug approval administration takes
many years to approve a drug for marketing. Owing to this complexity, the drug discovery
(Pascoalino et al;2016), development and approval becomes a risky process in the scientific
industry, and it impends a major challenge to the biomedical industry. Much of the risks
leading to failures, account for almost about seventeen per cent of total research
‘development costs’.
5

The mentioned monetary failures are expensive and the ‘body of knowledge’ dealing
with disease control, are held responsible for it. Research institutions and academic health
institutions play a critical role in analyzing, re-analyzing and demonstrating the targets that
can be applied to small molecules, in order to carry out the much-required clinical trials. For
therapeutic proteins (Lagassé et al;2017), the procedure involving discovery and
development followed by an approval is equally a lengthy and critical process. Even after
this, the market success of these biologics is uncertain. Biologics are extracted from the living
sources that the animals, viruses and bacteria. Then, these products like vaccines, monoclonal
antibodies are developed under regulations of FDA. The academic health institutions and the
research centers have produced various biological agents, in collaboration with biotechnology
and pharmaceutical companies.
Medical devices and equipment involves a spectrum of technologies starting from
disposable surgical gloves, thermometers and syringes to sophisticated imaging equipment,
prosthetics, angiographic stents, biomedical devices etc. Reflecting on the diversity, which is
the path circumscribed around ‘conception of an idea’ to ‘product development’ of these
biologics and drugs (LeBeau 2019). Involveent of academic, government researchers and
non-governmental organisations is critical for an accurate drug development process.
Premarket clearance or a premarket approval must be obtained from FDA before marketing a
medical device or a drug.
Medical devices and drugs are always checked for efficacy and safety. The European
Union and the United States approach faces a completely different set of challenges in these
aspects. Whereas the United States strictly relies on a centralized process acting through the
‘Food and Drug Administration also known as FDA’, ‘European Commission (EU)’ on the
other hand – have synchronised regulations inspired by norms of twenty-eight countries that
was combined to form the ‘European Union’. Historically, FDA was a consumer protection
6
with disease control, are held responsible for it. Research institutions and academic health
institutions play a critical role in analyzing, re-analyzing and demonstrating the targets that
can be applied to small molecules, in order to carry out the much-required clinical trials. For
therapeutic proteins (Lagassé et al;2017), the procedure involving discovery and
development followed by an approval is equally a lengthy and critical process. Even after
this, the market success of these biologics is uncertain. Biologics are extracted from the living
sources that the animals, viruses and bacteria. Then, these products like vaccines, monoclonal
antibodies are developed under regulations of FDA. The academic health institutions and the
research centers have produced various biological agents, in collaboration with biotechnology
and pharmaceutical companies.
Medical devices and equipment involves a spectrum of technologies starting from
disposable surgical gloves, thermometers and syringes to sophisticated imaging equipment,
prosthetics, angiographic stents, biomedical devices etc. Reflecting on the diversity, which is
the path circumscribed around ‘conception of an idea’ to ‘product development’ of these
biologics and drugs (LeBeau 2019). Involveent of academic, government researchers and
non-governmental organisations is critical for an accurate drug development process.
Premarket clearance or a premarket approval must be obtained from FDA before marketing a
medical device or a drug.
Medical devices and drugs are always checked for efficacy and safety. The European
Union and the United States approach faces a completely different set of challenges in these
aspects. Whereas the United States strictly relies on a centralized process acting through the
‘Food and Drug Administration also known as FDA’, ‘European Commission (EU)’ on the
other hand – have synchronised regulations inspired by norms of twenty-eight countries that
was combined to form the ‘European Union’. Historically, FDA was a consumer protection
6
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

agency while the ‘European Commission’s regulations’ were developed to bring a harmony
in state and nation’s commercial interests. Therefore, while the ‘FDA’ have a prior advantage
of centralization and ground rules – the European Union controls their medical device and
drug approval process through networks of both decentralized and centralised agencies
throughout its member states. There exists certain differences and similarities between ‘U.S
and Europe’s’ regulation of drugs and discusses challenges facing each. This study focusses
on the differences present between the United States and the European’s process of drug
approval.
There persists a discrepancy and controversy while differentiating between the
‘European Union and FDA’s regulatory processes can take both time and costs for a medical
drug and device to progress from the idea to an ‘approval’ under the given regulations. An
assertion process has been reported to be so slow that the FDA approval deprive the
American citizens of effective DADs. The critics have even characterized the FDA as being
slow, expensive and risk aversive. Additionally, the Institute of Medicine analysed that the
pre-marketing procedures of the FDA regarding medical devices is inadequate to ensure the
device safety, specifically of those that is approved largely due to their similarity with
‘predicate’ devices and not on a prospective, randomized clinical studies. The concern
associated with the European Union is that the DADs are approved very quickly which can
prove dangerous for the patient. In current years, there have been many advancements to
tighten the approval processes and to create an effective framework of regulatory policies
between the EU and the FDA (Davit et al; 2016). Efforts involve a recent legislation
proposed by U.S. Congress in order to facilitate a rapid release of drugs in the United States
that are already approved by the European Union.
7
in state and nation’s commercial interests. Therefore, while the ‘FDA’ have a prior advantage
of centralization and ground rules – the European Union controls their medical device and
drug approval process through networks of both decentralized and centralised agencies
throughout its member states. There exists certain differences and similarities between ‘U.S
and Europe’s’ regulation of drugs and discusses challenges facing each. This study focusses
on the differences present between the United States and the European’s process of drug
approval.
There persists a discrepancy and controversy while differentiating between the
‘European Union and FDA’s regulatory processes can take both time and costs for a medical
drug and device to progress from the idea to an ‘approval’ under the given regulations. An
assertion process has been reported to be so slow that the FDA approval deprive the
American citizens of effective DADs. The critics have even characterized the FDA as being
slow, expensive and risk aversive. Additionally, the Institute of Medicine analysed that the
pre-marketing procedures of the FDA regarding medical devices is inadequate to ensure the
device safety, specifically of those that is approved largely due to their similarity with
‘predicate’ devices and not on a prospective, randomized clinical studies. The concern
associated with the European Union is that the DADs are approved very quickly which can
prove dangerous for the patient. In current years, there have been many advancements to
tighten the approval processes and to create an effective framework of regulatory policies
between the EU and the FDA (Davit et al; 2016). Efforts involve a recent legislation
proposed by U.S. Congress in order to facilitate a rapid release of drugs in the United States
that are already approved by the European Union.
7

1.2 Problem statement
In general, the EMA and the FDA have objectives: a) promotion of public health b)
assess the efficacy and safety of the therapeutic products. c) Collaboration with the experts in
order to enrich the product development process. Both the authorities mandate a preclinical
testing that involve three-phased clinical trials and the final approval procedure. These are
integral parts of drug development. In the United States, the clinical trials and the market
approval are taken under supervision of the FDA (Dowell et al; 2016). In European Union,
the clinical trials (Eder and Wild 2019) can be started by member states and then, the market
authorizations can initiate a decentralized, centralized or a mutually recognized pathway.
Centralized pathway permits a proposed drug to be scrutinized by the ‘EMA’ and then to be
recommended to ‘the European Commission’ for an ultimate approval.
Market approval within the framework of the European Union becomes complex by
the extra regulations adopted by different member states. All these regulations ultimately
decide whether a drug can be marketed in a specific state or not. A drug that is already
approved by the EMA also needs an approval from the Medicines and Healthcare Products
Regulatory Agency in order to be finally marketed in United Kingdom (Davis et al; 2017).
Additionally, the National Institute for Health and Care Excellence assess the cardinal cost
concerns to understand if National Health Service shall purchase the same drug for patients’
application. Finally, each of the European Union member states supervise and regulate the
‘promotion and sale activities’ of all the pharmaceuticals. Consequently, National regulatory
authorities become responsible for regulation of pharmaceutical advertising (Koiniget et al;
2018), comparatively less restrictive than the United States.
The primary issues that affects the drug approval and development in international
markets - is the cost related to requirements of more unanticipated trials that can finally
increase the end price of the drug. Any sort of delays in the review process can reduce the
8
In general, the EMA and the FDA have objectives: a) promotion of public health b)
assess the efficacy and safety of the therapeutic products. c) Collaboration with the experts in
order to enrich the product development process. Both the authorities mandate a preclinical
testing that involve three-phased clinical trials and the final approval procedure. These are
integral parts of drug development. In the United States, the clinical trials and the market
approval are taken under supervision of the FDA (Dowell et al; 2016). In European Union,
the clinical trials (Eder and Wild 2019) can be started by member states and then, the market
authorizations can initiate a decentralized, centralized or a mutually recognized pathway.
Centralized pathway permits a proposed drug to be scrutinized by the ‘EMA’ and then to be
recommended to ‘the European Commission’ for an ultimate approval.
Market approval within the framework of the European Union becomes complex by
the extra regulations adopted by different member states. All these regulations ultimately
decide whether a drug can be marketed in a specific state or not. A drug that is already
approved by the EMA also needs an approval from the Medicines and Healthcare Products
Regulatory Agency in order to be finally marketed in United Kingdom (Davis et al; 2017).
Additionally, the National Institute for Health and Care Excellence assess the cardinal cost
concerns to understand if National Health Service shall purchase the same drug for patients’
application. Finally, each of the European Union member states supervise and regulate the
‘promotion and sale activities’ of all the pharmaceuticals. Consequently, National regulatory
authorities become responsible for regulation of pharmaceutical advertising (Koiniget et al;
2018), comparatively less restrictive than the United States.
The primary issues that affects the drug approval and development in international
markets - is the cost related to requirements of more unanticipated trials that can finally
increase the end price of the drug. Any sort of delays in the review process can reduce the
8

lucrative use of a patent and then, pointlessly limit the treatment choices for the patients with
a progressive disease or terminal pathology. The centralized authorization for oncology
therapeutics (Jonsson et al; 2017), in the European Union that involve many regulatory
entities like the European Commission, the EMA and its members states – takes twice as
much the approval time longthan in the United states.
Despite, submission of the identical data in support of the same drug, the FDA and the
EMA have reached dissimilar conclusions and evaluations (Jasińska-Stroschein et al; 2017).
Between the year of 1995 and the year of 2008, the EMA or the FDA approved twenty
percent of the oncological pharmaceuticals but twenty eight percent of the approved drugs
used a varied label wording. Likewise, a review of the existing drugs can cause restrictive
actions. Orlaam as a drug was found to have no proven life enhancing effects over the years
but ‘the FDA’ continued the drug in the United States market instead of instructing labelling
revisions. The manufacturer eventually withdrew Orlaam in the year of 2003 when drug sales
decreased dramatically due to the ‘FDA warnings’ (Sifuentes and Giuffrida, 2015).
Discrepancies in product categorization for the review presents a barrier. The ‘FDA’ and the
‘EMA’ have various standards for ‘labelling’ (Paglialunga et al; 2017) of products like
cosmetics or drugs, resulting in inconsistent obtainability for the consumer – ‘eight sunscreen
constituents’ are approved for a use in Europe were languished under FDA review for about
ten years and more. In 2014, advocates and manufacturers claimed that their sunscreens
provide suitable protection against the ‘sun damage’ and the ‘United States Congress’ then
passed a new ‘Sunscreen Innovation Act’ in order to accelerate review process of sunscreen
products. In about seven months, ‘all these eight applications’ were rejected due to its safety
‘non-complaice.’ The inconsistencies’ raise due to the fact that, the ‘EMA categorizes the
sunscreens as being cosmetics, whereas the FDA reviews it as over-the-counter drugs –
requiring the evidence for both the safety and its efficacy (Ozsvári et al; 2016). Because of a
9
a progressive disease or terminal pathology. The centralized authorization for oncology
therapeutics (Jonsson et al; 2017), in the European Union that involve many regulatory
entities like the European Commission, the EMA and its members states – takes twice as
much the approval time longthan in the United states.
Despite, submission of the identical data in support of the same drug, the FDA and the
EMA have reached dissimilar conclusions and evaluations (Jasińska-Stroschein et al; 2017).
Between the year of 1995 and the year of 2008, the EMA or the FDA approved twenty
percent of the oncological pharmaceuticals but twenty eight percent of the approved drugs
used a varied label wording. Likewise, a review of the existing drugs can cause restrictive
actions. Orlaam as a drug was found to have no proven life enhancing effects over the years
but ‘the FDA’ continued the drug in the United States market instead of instructing labelling
revisions. The manufacturer eventually withdrew Orlaam in the year of 2003 when drug sales
decreased dramatically due to the ‘FDA warnings’ (Sifuentes and Giuffrida, 2015).
Discrepancies in product categorization for the review presents a barrier. The ‘FDA’ and the
‘EMA’ have various standards for ‘labelling’ (Paglialunga et al; 2017) of products like
cosmetics or drugs, resulting in inconsistent obtainability for the consumer – ‘eight sunscreen
constituents’ are approved for a use in Europe were languished under FDA review for about
ten years and more. In 2014, advocates and manufacturers claimed that their sunscreens
provide suitable protection against the ‘sun damage’ and the ‘United States Congress’ then
passed a new ‘Sunscreen Innovation Act’ in order to accelerate review process of sunscreen
products. In about seven months, ‘all these eight applications’ were rejected due to its safety
‘non-complaice.’ The inconsistencies’ raise due to the fact that, the ‘EMA categorizes the
sunscreens as being cosmetics, whereas the FDA reviews it as over-the-counter drugs –
requiring the evidence for both the safety and its efficacy (Ozsvári et al; 2016). Because of a
9
Secure Best Marks with AI Grader
Need help grading? Try our AI Grader for instant feedback on your assignments.

wide gap in manufacturing costs of cosmetics and the drugs, this divergent approach by two
regulatory agencies signify a challenging obstacle that prevents market expansion. Similar
issues also ‘affects’ the grey areas lying between the cosmetics and the therapeutics including
dental, hair and skin care products.
One of the most crucial steps in drug development is obtaining the marketing
approval of a new agent. After approval, the agents are then, prescribed to the patients that
follow a labeled indication. Traditionally, cancer drugs are prescribed on a histological basis,
line of therapy and stage of the disease. With molecular understanding of cancer progression
process (metastasis), the medicines prescribed on a subgroup basis are critical and known as
companion or co - development diagnostics. In the principle, the enhancement strategies are
based on the predictive biomarkers (Jardim et al; 2015) that would lead to a smaller and
quicker, and less potentially expensive clinical trials, minimizing the exposure of patients to
ineffective therapies. Thus, it is crucial that sponsors, with the use of the IVD, identify the
subpopulation that will benefit the most with the new investigational agent. In this review, we
attempt to provide a brief overview and comparison of the oncology drug approval process in
the United States and the European Union (EU). To illustrate similarities and differences
between regions, we discuss three interesting submissions with distinct outcomes in the
United States and the EU. Moreover, we describe the main differences in the approval
process for drugs that require an IVD. This review focuses on the approval requirements for
late oncology development. Early-stage development of oncology drugs has been previously
addressed elsewhere. This review is divided into five sections: preapproval (or premarketing)
interactions with regulatory agencies, approval procedures (or marketing interactions),
companion diagnostics, expedited programs, and examples of recent drug approvals.
10
regulatory agencies signify a challenging obstacle that prevents market expansion. Similar
issues also ‘affects’ the grey areas lying between the cosmetics and the therapeutics including
dental, hair and skin care products.
One of the most crucial steps in drug development is obtaining the marketing
approval of a new agent. After approval, the agents are then, prescribed to the patients that
follow a labeled indication. Traditionally, cancer drugs are prescribed on a histological basis,
line of therapy and stage of the disease. With molecular understanding of cancer progression
process (metastasis), the medicines prescribed on a subgroup basis are critical and known as
companion or co - development diagnostics. In the principle, the enhancement strategies are
based on the predictive biomarkers (Jardim et al; 2015) that would lead to a smaller and
quicker, and less potentially expensive clinical trials, minimizing the exposure of patients to
ineffective therapies. Thus, it is crucial that sponsors, with the use of the IVD, identify the
subpopulation that will benefit the most with the new investigational agent. In this review, we
attempt to provide a brief overview and comparison of the oncology drug approval process in
the United States and the European Union (EU). To illustrate similarities and differences
between regions, we discuss three interesting submissions with distinct outcomes in the
United States and the EU. Moreover, we describe the main differences in the approval
process for drugs that require an IVD. This review focuses on the approval requirements for
late oncology development. Early-stage development of oncology drugs has been previously
addressed elsewhere. This review is divided into five sections: preapproval (or premarketing)
interactions with regulatory agencies, approval procedures (or marketing interactions),
companion diagnostics, expedited programs, and examples of recent drug approvals.
10

1.3 Aims and Objectives
Aim – The study aims to compare the process of cancer drug approval by the ‘FDA’ and
‘EMA’.
Objectives
To explore the drug approval process of the Food and Drug administration (FDA)
To explore the drug approval process of the European Medicines Agency (EMA)
To explore the differences and similarities between operational processes of the FDA
and the EMA
1.4 Structure of the Research
Chapter One – the section describes the framework of the research. This chapter
involves a brief description of research background and it provides a rationale for the
research selection process. It comprises of aims and the objectives of the research.
Chapter Two – involves a literature review. Accordingly, analysis of the models and the
theoretical frameworks have. This chapter encompasses the terminology and arguments used
by other primary and secondary researches.
Chapter Three – addresses the methodology. Research process and research philosophy
are described in this chapter. Moreover, this chapter explains the research design. The data
collection methods, sampling aspects, along with the ethical considerations are included in
this chapter. Data analysis of the collected data is done in this section.
Chapter Four – Constitutes the results and discussions. This section plays a crucial role in
achieving of the research objectives and objectives.
11
Aim – The study aims to compare the process of cancer drug approval by the ‘FDA’ and
‘EMA’.
Objectives
To explore the drug approval process of the Food and Drug administration (FDA)
To explore the drug approval process of the European Medicines Agency (EMA)
To explore the differences and similarities between operational processes of the FDA
and the EMA
1.4 Structure of the Research
Chapter One – the section describes the framework of the research. This chapter
involves a brief description of research background and it provides a rationale for the
research selection process. It comprises of aims and the objectives of the research.
Chapter Two – involves a literature review. Accordingly, analysis of the models and the
theoretical frameworks have. This chapter encompasses the terminology and arguments used
by other primary and secondary researches.
Chapter Three – addresses the methodology. Research process and research philosophy
are described in this chapter. Moreover, this chapter explains the research design. The data
collection methods, sampling aspects, along with the ethical considerations are included in
this chapter. Data analysis of the collected data is done in this section.
Chapter Four – Constitutes the results and discussions. This section plays a crucial role in
achieving of the research objectives and objectives.
11

Chapter Five – provides a conclusion. The study précises the extent of research aim
achievement. This chapter reveals the limitations of research study and then highlights the
scope of future researches in similar areas.
Chapter 2 – Literature review
Introduction
Much before any new prescription medicines are permitted into the public market, they
are tested through critical studies and whether the drug regulation agencies are satisfied, that
whether the benefits of the drug outweigh dangers of toxicity (Shields et al; 2016). The most
valuable and informative studies provides randomized controlled trials mediated evidences on
whether the new drug provides the outcomes, significant to patients and whether the results
can be replicated over different populations in comparison with different treatment options.
Lastly and most importantly – the results or the outcomes are ‘scrutinized’ for being
clinically meaningful (Prigerson et al; 2018). The goal of oncological treatment is improving
the duration and quality of life. Clinical trials are intended to increase the chances of
regulatory approval for fresh drugs that assesses indirect measures of a drug efficacy.
According to the FDA perspective, in the year of 2012, the Food and Drug
Administration Safety and Innovation Act made it sound that patient-centered drug
development has to be priority for the Congress, ‘American people’ and the ‘FDA’. There has
been a little research done about the ‘patient’s symptoms’ roused by the injected drugs and
this can prevented and managed by direct measures indeed. The challenge, always the
challenge lies in how the ‘patient’s perspective’ can be accurately captured. The FDA need a
substantial evidence to be presented in support of labeling and a marketing claim that benefits
and necessities are adequate and in controlled studies that used as a set of ‘reliable’
assessments. While evaluating if PRO tool is adequate for support labeling, this agency then
12
achievement. This chapter reveals the limitations of research study and then highlights the
scope of future researches in similar areas.
Chapter 2 – Literature review
Introduction
Much before any new prescription medicines are permitted into the public market, they
are tested through critical studies and whether the drug regulation agencies are satisfied, that
whether the benefits of the drug outweigh dangers of toxicity (Shields et al; 2016). The most
valuable and informative studies provides randomized controlled trials mediated evidences on
whether the new drug provides the outcomes, significant to patients and whether the results
can be replicated over different populations in comparison with different treatment options.
Lastly and most importantly – the results or the outcomes are ‘scrutinized’ for being
clinically meaningful (Prigerson et al; 2018). The goal of oncological treatment is improving
the duration and quality of life. Clinical trials are intended to increase the chances of
regulatory approval for fresh drugs that assesses indirect measures of a drug efficacy.
According to the FDA perspective, in the year of 2012, the Food and Drug
Administration Safety and Innovation Act made it sound that patient-centered drug
development has to be priority for the Congress, ‘American people’ and the ‘FDA’. There has
been a little research done about the ‘patient’s symptoms’ roused by the injected drugs and
this can prevented and managed by direct measures indeed. The challenge, always the
challenge lies in how the ‘patient’s perspective’ can be accurately captured. The FDA need a
substantial evidence to be presented in support of labeling and a marketing claim that benefits
and necessities are adequate and in controlled studies that used as a set of ‘reliable’
assessments. While evaluating if PRO tool is adequate for support labeling, this agency then
12
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

emphasizes the ‘content validity’ that this tool assesses for an intended purpose (Wallwiener
et al; 2017). No recent researches have systematically inspected the evidences or the
magnitude in benefit of cancer drugs improvement (and approval) by the European Medicines
Agency.
Available data in ‘the United States’ has shown that the ‘Food and Drug Administration
approve a ‘small proportion of the oncological treatments’. In addition, it has drastically
affected the survival and quality of life (Brunault et al; 2017). FDA- the EMA comparative
studies has shown differences in the regulatory decisions, the approval routes, and oncologic
drug approvals that have vital implications for a patients’ safety and for clinical practices.
2.2 Review
Arnold et al., 2016 aimed to study the role of a Patient-Reported Outcome Evaluation
in the Approval of the ‘Orphan Drugs. Theyreviewed 15 years of drugs approvals by ‘FDA
and EMA’. In 2016, the International Rare Diseases Research Consortium (Austin et al;
2017) concluded that ‘the patient-reported outcome measures’ is important in the clinical
researches of ‘critical cancer conditions’ as they assess the main treatment benefits from the
patient’s perspective. The focus of the study is ‘an evaluation of the ‘Food and Drug
Administration and the EMA’ use of PROs while evaluating orphan drugs. The methods – the
‘EMA’ and the ‘FDA websites’ was ‘reviewed by the study’ in order to retrieve the products
sanctioned by orphan designation from ‘January 2002 to July 2017’. The ‘FDA label’ and
‘summary of the EMA’ ‘product characteristics’ were analyzed to ‘discover’ the ‘use of
‘PROs’. According to the study results - a research review of both the ‘EMA’ and ‘FDA’
(with exclusion of the duplicate records), led to ‘452’ distinct designations. The designations
for the products that is indicated for an oncology purpose, was excluded by the study and this
left ‘258’ designations to review. Only about ‘45’ designations included a PRO claims in
their labeling, that is ‘17.4%’ of the total designations of non-oncology products. Forty-two
13
et al; 2017). No recent researches have systematically inspected the evidences or the
magnitude in benefit of cancer drugs improvement (and approval) by the European Medicines
Agency.
Available data in ‘the United States’ has shown that the ‘Food and Drug Administration
approve a ‘small proportion of the oncological treatments’. In addition, it has drastically
affected the survival and quality of life (Brunault et al; 2017). FDA- the EMA comparative
studies has shown differences in the regulatory decisions, the approval routes, and oncologic
drug approvals that have vital implications for a patients’ safety and for clinical practices.
2.2 Review
Arnold et al., 2016 aimed to study the role of a Patient-Reported Outcome Evaluation
in the Approval of the ‘Orphan Drugs. Theyreviewed 15 years of drugs approvals by ‘FDA
and EMA’. In 2016, the International Rare Diseases Research Consortium (Austin et al;
2017) concluded that ‘the patient-reported outcome measures’ is important in the clinical
researches of ‘critical cancer conditions’ as they assess the main treatment benefits from the
patient’s perspective. The focus of the study is ‘an evaluation of the ‘Food and Drug
Administration and the EMA’ use of PROs while evaluating orphan drugs. The methods – the
‘EMA’ and the ‘FDA websites’ was ‘reviewed by the study’ in order to retrieve the products
sanctioned by orphan designation from ‘January 2002 to July 2017’. The ‘FDA label’ and
‘summary of the EMA’ ‘product characteristics’ were analyzed to ‘discover’ the ‘use of
‘PROs’. According to the study results - a research review of both the ‘EMA’ and ‘FDA’
(with exclusion of the duplicate records), led to ‘452’ distinct designations. The designations
for the products that is indicated for an oncology purpose, was excluded by the study and this
left ‘258’ designations to review. Only about ‘45’ designations included a PRO claims in
their labeling, that is ‘17.4%’ of the total designations of non-oncology products. Forty-two
13

different products were represented of which 10 were common to both agencies. PRO
measures were cited in label that cardinally focused on symptoms - dyspnea, pain and fatigue.
It rarely focused on the health-related functionality of life. In cases of CAPS, acromegaly
(Melmed 2017) and cystic fibrosis (Wainwright et al; 2015) - the measures taken were very
specific to the rare conditions. The study ‘concluded’ that the ‘review’ revealed a patient’s
perspective while evaluating the orphan drugs – was not properly implemented. A number of
rare diseases prevalent in small patient numbers for each of the explicit pathology, a lack of
knowledge about the natural history regarding diseases or an association of intellectual and
disability impairments make PRO development a difficult task.
Zeitoun et al; (2017) aimed to understand the post-marketing studies for the novel
drugs, approved by ‘the EMA’ and the ‘FDA’, between ‘the year of 2010 and 2005’ through
a ‘cross-sectional study’. The study had the ‘objective’ to ‘characterize the post-marketing
studies of drugs’, which were already ‘approved’ by the ‘US FDA and EMA’. In the ‘Design
and setting’ – ‘ClinicalTrials.gov’ performed ‘clinical trials’ until September of 2014 and
focussed from ‘2005 to 2010’. The Regulatory documents in both agencies were used.The
secondary and primary outcome were then focussed. All the identified post-marketing studies
were then classified according to the strategies of enrolment, status, funds, and the
geographical locations. The research determined whether the studies actually analysed the
first approved indication. In the findings – it was found that about ‘69’ novel drugs were
approved between the year of 2005 and 2010. A total of ‘6679’ post-marketing studies
(relevant) were then identified of which - 5972 was found to be interventional. A median
studies per drug were ‘50’and the median number per study were ‘60’. Overall, 2901 studies
were fully completed, 487 were terminated, 1013 were active but not recruiting, 1895 were
recruiting and 319 were not recruiting. About ‘80%’ of the studies ‘were’ conducted in
‘Europe’ and ‘North America’, ‘2441’ were studied under a different indication of the
14
measures were cited in label that cardinally focused on symptoms - dyspnea, pain and fatigue.
It rarely focused on the health-related functionality of life. In cases of CAPS, acromegaly
(Melmed 2017) and cystic fibrosis (Wainwright et al; 2015) - the measures taken were very
specific to the rare conditions. The study ‘concluded’ that the ‘review’ revealed a patient’s
perspective while evaluating the orphan drugs – was not properly implemented. A number of
rare diseases prevalent in small patient numbers for each of the explicit pathology, a lack of
knowledge about the natural history regarding diseases or an association of intellectual and
disability impairments make PRO development a difficult task.
Zeitoun et al; (2017) aimed to understand the post-marketing studies for the novel
drugs, approved by ‘the EMA’ and the ‘FDA’, between ‘the year of 2010 and 2005’ through
a ‘cross-sectional study’. The study had the ‘objective’ to ‘characterize the post-marketing
studies of drugs’, which were already ‘approved’ by the ‘US FDA and EMA’. In the ‘Design
and setting’ – ‘ClinicalTrials.gov’ performed ‘clinical trials’ until September of 2014 and
focussed from ‘2005 to 2010’. The Regulatory documents in both agencies were used.The
secondary and primary outcome were then focussed. All the identified post-marketing studies
were then classified according to the strategies of enrolment, status, funds, and the
geographical locations. The research determined whether the studies actually analysed the
first approved indication. In the findings – it was found that about ‘69’ novel drugs were
approved between the year of 2005 and 2010. A total of ‘6679’ post-marketing studies
(relevant) were then identified of which - 5972 was found to be interventional. A median
studies per drug were ‘50’and the median number per study were ‘60’. Overall, 2901 studies
were fully completed, 487 were terminated, 1013 were active but not recruiting, 1895 were
recruiting and 319 were not recruiting. About ‘80%’ of the studies ‘were’ conducted in
‘Europe’ and ‘North America’, ‘2441’ were studied under a different indication of the
14

approved condition. The ‘studies’ that were approved indicated that industries have
sponsored more than ‘68.7% versus 53.7%’ with a p value of less than ‘0.0001’. The post-
marketing pharmaceutical studies were reported to be highly variable and that it
predominantly spanned over Europe and North America. Some questions were reassuring but
the majority lacked a sense of coordination in post-marketing research. Elderly patients, who
were receiving the anticancer drugs, were suspected to have an elevated risk to treatment
toxicities as compared to others – the younger peers. Pharmacokinetic underpinnings of
elevated risk was constantly assessed.
Rose et al; (2016) aimed to study the pharmacokinetics of selected anticancer drugs in
elderly cancer patients. They focused their study on breast cancer. The elderly patients were
given ‘oncological drugs’, which can have elevated risks to produce ‘treatment toxicity’ as
compared to the ‘younger peers’. The aim of the study was analysing the effect of age on
anticancer agent’s pharmacokinetics – used more frequently in breast cancer patients. As for
processes: they searched through PubMed electronic database, SmPC and drug approval
reviews done by the ‘FDA’ and the ‘EMA’. The various publications, which explain age
related pharmacokinetic profiles, involved in anticancer drug development against breast
cancer that excluded the endocrine compounds, were then selected. The review provides an
overview of available data that then explain the impact of an increasing age on anticancer
drugs’ pharmacokinetics used for the cure of breast cancer. The selected and published data
then revealed that the effect and increasing age were differentially associated with
pharmacokinetics of the oncological drugs. These age-related pharmacokinetic differences of
anthracyclines and the platina agents are of clinical importance. In the majority of cases, the
age cannot be regarded as a reliable marker for the anticancer drugs pharmacokinetics. The
study emphasized on taking of a geriatric assessment (functional) before prescription of the
drugs.
15
sponsored more than ‘68.7% versus 53.7%’ with a p value of less than ‘0.0001’. The post-
marketing pharmaceutical studies were reported to be highly variable and that it
predominantly spanned over Europe and North America. Some questions were reassuring but
the majority lacked a sense of coordination in post-marketing research. Elderly patients, who
were receiving the anticancer drugs, were suspected to have an elevated risk to treatment
toxicities as compared to others – the younger peers. Pharmacokinetic underpinnings of
elevated risk was constantly assessed.
Rose et al; (2016) aimed to study the pharmacokinetics of selected anticancer drugs in
elderly cancer patients. They focused their study on breast cancer. The elderly patients were
given ‘oncological drugs’, which can have elevated risks to produce ‘treatment toxicity’ as
compared to the ‘younger peers’. The aim of the study was analysing the effect of age on
anticancer agent’s pharmacokinetics – used more frequently in breast cancer patients. As for
processes: they searched through PubMed electronic database, SmPC and drug approval
reviews done by the ‘FDA’ and the ‘EMA’. The various publications, which explain age
related pharmacokinetic profiles, involved in anticancer drug development against breast
cancer that excluded the endocrine compounds, were then selected. The review provides an
overview of available data that then explain the impact of an increasing age on anticancer
drugs’ pharmacokinetics used for the cure of breast cancer. The selected and published data
then revealed that the effect and increasing age were differentially associated with
pharmacokinetics of the oncological drugs. These age-related pharmacokinetic differences of
anthracyclines and the platina agents are of clinical importance. In the majority of cases, the
age cannot be regarded as a reliable marker for the anticancer drugs pharmacokinetics. The
study emphasized on taking of a geriatric assessment (functional) before prescription of the
drugs.
15
Secure Best Marks with AI Grader
Need help grading? Try our AI Grader for instant feedback on your assignments.

Vivot et al; (2017) aimed to understand the price, clinical benefits and the approval
features of the FDA. The study ‘focussed’ on the new approved drugs for the treatment of a
progressive solid cancer. In the years of 2000 to 2015 – the prices of oncologic drugs was
found to be increasing. This study then attempted to draw a correlation between the price and
the clinical uses of the oncologic drugs. The study analyses the molecular entities, the new
biologics while treating advanced cancer (solid). Then the clinical benefits was graded
according to medical review of the FDA clinical trials of 2016. The findings are in
accordance with the American Society of Clinical Oncology Value Framework (Schnipper et
al; 2016) and with the European Society for Medical Oncology Magnitude of Clinical
Benefit Scale. Features of approvals and drugs were taken from the publicly obtainable the
FDA documents and the price was then evaluated in accordance with the ‘United States
Medicare’. According to the research study, the FDA approved about ‘51’ new drugs to treat
the advanced stages of solid cancer in 2000 through to 2015. According to the ESMO-MCBS,
about ‘5’ drugs were rated as grade one which is the lowest, ‘9’ of the drugs are grade two,
‘10’ of them were grade ‘three’, ‘11’of them were grade four and 2 were found to be at grade
five which is the highest. Thirteen drugs revealed a meaningful clinical benefit. According to
the ASCO-VF - the drug value (median) was found to be ‘37’. No relationship was
established between drug price and clinical benefits. The study concluded that newly FDA-
approved oncologic drugs do not have increased clinical advantages as assessed by the
current scales. No important relation was found between price of the oncologic drugs and its
benefits to patients and society.
Davis et al; (2017) aims to understand the availability of the evidence on benefits of
overall survival, the quality of life improved by oncologic drugs as approved by the European
Medicines Agency. The study ‘performed’ a cohort study or retrospective on drug approvals
between 2009 and 2013. The study used a publicly accessible setting of regulatory scientific
16
features of the FDA. The study ‘focussed’ on the new approved drugs for the treatment of a
progressive solid cancer. In the years of 2000 to 2015 – the prices of oncologic drugs was
found to be increasing. This study then attempted to draw a correlation between the price and
the clinical uses of the oncologic drugs. The study analyses the molecular entities, the new
biologics while treating advanced cancer (solid). Then the clinical benefits was graded
according to medical review of the FDA clinical trials of 2016. The findings are in
accordance with the American Society of Clinical Oncology Value Framework (Schnipper et
al; 2016) and with the European Society for Medical Oncology Magnitude of Clinical
Benefit Scale. Features of approvals and drugs were taken from the publicly obtainable the
FDA documents and the price was then evaluated in accordance with the ‘United States
Medicare’. According to the research study, the FDA approved about ‘51’ new drugs to treat
the advanced stages of solid cancer in 2000 through to 2015. According to the ESMO-MCBS,
about ‘5’ drugs were rated as grade one which is the lowest, ‘9’ of the drugs are grade two,
‘10’ of them were grade ‘three’, ‘11’of them were grade four and 2 were found to be at grade
five which is the highest. Thirteen drugs revealed a meaningful clinical benefit. According to
the ASCO-VF - the drug value (median) was found to be ‘37’. No relationship was
established between drug price and clinical benefits. The study concluded that newly FDA-
approved oncologic drugs do not have increased clinical advantages as assessed by the
current scales. No important relation was found between price of the oncologic drugs and its
benefits to patients and society.
Davis et al; (2017) aims to understand the availability of the evidence on benefits of
overall survival, the quality of life improved by oncologic drugs as approved by the European
Medicines Agency. The study ‘performed’ a cohort study or retrospective on drug approvals
between 2009 and 2013. The study used a publicly accessible setting of regulatory scientific
16

reports on the oncologic approvals by the European Medicines Agency between 2009 and
2013. As outcome measures, the post-marketing pivotal trials of the oncologic drugs were
taken, in accordance with the randomisation, blinding, crossover, comparators, endpoints.
The availability and the magnitude of the benefits of the overall survival of the quality of life
during approval time and ‘post-market’ entry. The Validated European Society for Medical
Oncology Magnitude of Clinical Benefit Scale (ESMO-MCBS) used to assess the clinical
value of the reported gains in published studies of cancer drugs. The study showed that
between the 2009 and 2013- during the market approval, there was an improvement in the
‘overall quality’ of life in 7 out of 68 indications. Of forty four indications- ‘7’ revealed an
evidence of extension of life. Eleven per cent reported a benefit on the quality of life. In 68
cancer cases with the EMA approval with a median follow up of 5.4 years, ‘just 51%’
revealed a considerable improvement of quality of life. Thirty three per cent was found to be
uncertain. Of about 23 indications, related to survival benefit, less than half were proved to be
clinically meaningful. The study concluded that the systematic evaluation of the cancer drug
approvals by the EMA from the year of 2009 through to 2013 reveals that most of the drugs
gets into the market and this is without much evidence of improved quality of life. After 3
and half years of market entry, not much conclusive evidence was found regarding these
drugs and that, these improved the overall quality of life in cancer patients.
Rose (2019) aimed to study the pediatric cancer studies as started by the United
States’ food and drug administration and the European medicines agency, which were aimed
at the labels, not at the better treatment. The study identifies that the young patients are
susceptible to mono-therapy rather than combination treatment (Kunz and Hölzel 2017). The
FDA started pediatric cancer studies in ‘United States’ and the ‘European medicines agency’
started the same, in Europe. The researchers focussed on one to ‘three FDA-triggered
paediatric oncology studies (Wdlff 1991) in the literature and on ‘the EMA/FDA paediatric
17
2013. As outcome measures, the post-marketing pivotal trials of the oncologic drugs were
taken, in accordance with the randomisation, blinding, crossover, comparators, endpoints.
The availability and the magnitude of the benefits of the overall survival of the quality of life
during approval time and ‘post-market’ entry. The Validated European Society for Medical
Oncology Magnitude of Clinical Benefit Scale (ESMO-MCBS) used to assess the clinical
value of the reported gains in published studies of cancer drugs. The study showed that
between the 2009 and 2013- during the market approval, there was an improvement in the
‘overall quality’ of life in 7 out of 68 indications. Of forty four indications- ‘7’ revealed an
evidence of extension of life. Eleven per cent reported a benefit on the quality of life. In 68
cancer cases with the EMA approval with a median follow up of 5.4 years, ‘just 51%’
revealed a considerable improvement of quality of life. Thirty three per cent was found to be
uncertain. Of about 23 indications, related to survival benefit, less than half were proved to be
clinically meaningful. The study concluded that the systematic evaluation of the cancer drug
approvals by the EMA from the year of 2009 through to 2013 reveals that most of the drugs
gets into the market and this is without much evidence of improved quality of life. After 3
and half years of market entry, not much conclusive evidence was found regarding these
drugs and that, these improved the overall quality of life in cancer patients.
Rose (2019) aimed to study the pediatric cancer studies as started by the United
States’ food and drug administration and the European medicines agency, which were aimed
at the labels, not at the better treatment. The study identifies that the young patients are
susceptible to mono-therapy rather than combination treatment (Kunz and Hölzel 2017). The
FDA started pediatric cancer studies in ‘United States’ and the ‘European medicines agency’
started the same, in Europe. The researchers focussed on one to ‘three FDA-triggered
paediatric oncology studies (Wdlff 1991) in the literature and on ‘the EMA/FDA paediatric
17

reports’. According to the results, the authors of FDA focussed on two primary assumptions
which are - (1) Children, less than seventeen years, need a separate proof for efficacy. (2)
Children with myelogenous leukaemia (chronic) where the pathophysiology happens to be
distinct from an adult cancer (Hochhaus et al;2017). The FDA-led studies used single
cytotoxic agents in refractory or relapsed patient less than 21 years of age (Saber and
Leighton 2015). Nowadays, combination treatment can be up to thirteen cytotoxic agents in a
standardized oncology care. Any treatment with single session of chemotherapeutic agent
was reported not to increase survival longevity of the patients. The ‘European Union’
expanded in order to provide the children under 18 years of age with ‘paediatric
interventions’ plans in the rarest of cancer conditions. One of the FDA-mediated package
examined the effect of ipilimumab in the ‘paediatric’ melanoma (Strouse 2005), and thirteen
EMA PIP ‘paediatric’ studies in the solid tumours including the melanoma. Two
monotherapy (paediatric) studies with vemurafenib and ipilimumab were terminated in the
year of 2016 while five others kept on recruiting. A study on ‘FDA/EMA’ on ‘paediatric’
oncology focussed on treatment of children. According to the study, the FDA assumptions
that encompass a different biology about ‘paediatric’ malignancy is mostly incorrect. The’ ‘
FDA/EMA’reported to provide the list regulatory and this list is not comprised of any
therapeutic achievements. ‘European Union’ researchers then performed the oncology studies
predominantly in order to come up with new drug plans. EMA studies on ‘medications’ focs
on life-saving drugs and the threats posed by them (Ades et al;2017). The study concludes
that the FDA representatives sometimes augmented the concept of ‘therapeutic orphans’ by
making incorrect assumptions regarding the ‘paediatric’ malignancies (Gammie et al, 2015).
The European medical agency then further expanded on ‘Paediatric Imperative’ (Rodieux et
al;2019). That study confirmed that PIPs does not provide beneficial treatment. The study
implies that the ethical committees should re-analyse the ongoing ‘paediatric oncology’
18
which are - (1) Children, less than seventeen years, need a separate proof for efficacy. (2)
Children with myelogenous leukaemia (chronic) where the pathophysiology happens to be
distinct from an adult cancer (Hochhaus et al;2017). The FDA-led studies used single
cytotoxic agents in refractory or relapsed patient less than 21 years of age (Saber and
Leighton 2015). Nowadays, combination treatment can be up to thirteen cytotoxic agents in a
standardized oncology care. Any treatment with single session of chemotherapeutic agent
was reported not to increase survival longevity of the patients. The ‘European Union’
expanded in order to provide the children under 18 years of age with ‘paediatric
interventions’ plans in the rarest of cancer conditions. One of the FDA-mediated package
examined the effect of ipilimumab in the ‘paediatric’ melanoma (Strouse 2005), and thirteen
EMA PIP ‘paediatric’ studies in the solid tumours including the melanoma. Two
monotherapy (paediatric) studies with vemurafenib and ipilimumab were terminated in the
year of 2016 while five others kept on recruiting. A study on ‘FDA/EMA’ on ‘paediatric’
oncology focussed on treatment of children. According to the study, the FDA assumptions
that encompass a different biology about ‘paediatric’ malignancy is mostly incorrect. The’ ‘
FDA/EMA’reported to provide the list regulatory and this list is not comprised of any
therapeutic achievements. ‘European Union’ researchers then performed the oncology studies
predominantly in order to come up with new drug plans. EMA studies on ‘medications’ focs
on life-saving drugs and the threats posed by them (Ades et al;2017). The study concludes
that the FDA representatives sometimes augmented the concept of ‘therapeutic orphans’ by
making incorrect assumptions regarding the ‘paediatric’ malignancies (Gammie et al, 2015).
The European medical agency then further expanded on ‘Paediatric Imperative’ (Rodieux et
al;2019). That study confirmed that PIPs does not provide beneficial treatment. The study
implies that the ethical committees should re-analyse the ongoing ‘paediatric oncology’
18
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

studies by suspending the questionable ones and by rejecting the ‘inept’ ones. The study
declares that the EU and US paediatric laws requires revision.
2.3 Summary
Both the EMA and the FDA have similar and different gaps in the drug approval
process. There are discrepancies in both the operational processes. While the drugs are
approved very rapidly without much safety and risk assessments or the oncology drugs are
approved very late due to lack of infrastructures, analytic and resource issues. The oncology
drugs requires a great deal of care from discovery to designing to the final market approval
and any gap in any of the process excellence can create a difficult situation for both the
stakeholders and the clients. Having the correct guidelines, research philosophy and
appropriate chemical analysis (Desfontaine et al; 2015) at every level is very critical. Market
research, understanding the disease pathology and training the right workforce responsible for
designing the target drug is critical as well. While the ‘FDA’ and the ‘EMA’ have lacked in
certain areas such as planning, research, goal setting and operational processes over the years,
still FDA being a more established organization compared to the ‘EMA’, has been able to
understand the market needs better than the EMA. In terms of power and authority even, the
FDA has acquired more and more powers over the years while the EMA is still a subsidiary
of other big authorities, so when it comes to exercise of powers – the ‘FDA’ is placed in a
more authorized front as compared to the ‘EMA’. But there has been many lag in terms of
procedural development for oncology drug approval process on part of both EMA and FDA
(Cardot et al; 2016) Although there had been many differences between their working
policies and exercising of drug approval powers – there has been critical similarities between
their actions and executional ways. The drugs retained in the markets even after lacked
evidences to improve quality of life in cancer and other terminal patients. This is clearly due
to a lack of audit and reluctance in determining the real world outcomes of the released drugs.
19
declares that the EU and US paediatric laws requires revision.
2.3 Summary
Both the EMA and the FDA have similar and different gaps in the drug approval
process. There are discrepancies in both the operational processes. While the drugs are
approved very rapidly without much safety and risk assessments or the oncology drugs are
approved very late due to lack of infrastructures, analytic and resource issues. The oncology
drugs requires a great deal of care from discovery to designing to the final market approval
and any gap in any of the process excellence can create a difficult situation for both the
stakeholders and the clients. Having the correct guidelines, research philosophy and
appropriate chemical analysis (Desfontaine et al; 2015) at every level is very critical. Market
research, understanding the disease pathology and training the right workforce responsible for
designing the target drug is critical as well. While the ‘FDA’ and the ‘EMA’ have lacked in
certain areas such as planning, research, goal setting and operational processes over the years,
still FDA being a more established organization compared to the ‘EMA’, has been able to
understand the market needs better than the EMA. In terms of power and authority even, the
FDA has acquired more and more powers over the years while the EMA is still a subsidiary
of other big authorities, so when it comes to exercise of powers – the ‘FDA’ is placed in a
more authorized front as compared to the ‘EMA’. But there has been many lag in terms of
procedural development for oncology drug approval process on part of both EMA and FDA
(Cardot et al; 2016) Although there had been many differences between their working
policies and exercising of drug approval powers – there has been critical similarities between
their actions and executional ways. The drugs retained in the markets even after lacked
evidences to improve quality of life in cancer and other terminal patients. This is clearly due
to a lack of audit and reluctance in determining the real world outcomes of the released drugs.
19

The ‘safety and risk assessments of the drugs’ have been a seriously great issue but many
drugs which showed a good outcome in treatment of cancer subjects were also associated
with ‘safety isses’. Neither the ‘FDA’ nor the ‘EMA’ were conscious of the ‘unsafe and poor
outcomes’ even after these ‘cancer drugs’ are released on a ‘national level or a state level’.
Improper or lack of population studies while assessing the actual needs of a patient
population resulted in formulation of wrong or clinically irrelevant oncology drugs.
2.4 Literature gap
Most the previous studies have focused on the overall gaps in drug approval process
by the ‘FDA’ and the ‘EMA’. Many of the studiestried to understand the differences between
the ‘FDA and the EMA’ as well as their administrative and executional similarities. No
studies so far has researched about various possibilities of solving the problems with ‘FDA
and EMA’ with each other ‘strengths’. This study, other than revealing the new aspects of
similarities and differences with the ‘FDA’ and the ‘EMA’ – would also focus on the
research gap and propose a positive outcome based on the same.
Chapter 3 - Research methodology
3.1 Introduction
As, the study aims to compare the similarities and differences between ‘the FDA and
the EMA’, it takes up a descriptive approach to analyze the underpinnings of drug approval
process by both ‘the FDA and the EMA’. The methodology focuses on interpretation of the
secondary data. The data is both quantitative and qualitative. The study applies a reasoning to
comprehend the different unexamined aspects of oncology drug approval process, done by
EMA and FDA.
20
drugs which showed a good outcome in treatment of cancer subjects were also associated
with ‘safety isses’. Neither the ‘FDA’ nor the ‘EMA’ were conscious of the ‘unsafe and poor
outcomes’ even after these ‘cancer drugs’ are released on a ‘national level or a state level’.
Improper or lack of population studies while assessing the actual needs of a patient
population resulted in formulation of wrong or clinically irrelevant oncology drugs.
2.4 Literature gap
Most the previous studies have focused on the overall gaps in drug approval process
by the ‘FDA’ and the ‘EMA’. Many of the studiestried to understand the differences between
the ‘FDA and the EMA’ as well as their administrative and executional similarities. No
studies so far has researched about various possibilities of solving the problems with ‘FDA
and EMA’ with each other ‘strengths’. This study, other than revealing the new aspects of
similarities and differences with the ‘FDA’ and the ‘EMA’ – would also focus on the
research gap and propose a positive outcome based on the same.
Chapter 3 - Research methodology
3.1 Introduction
As, the study aims to compare the similarities and differences between ‘the FDA and
the EMA’, it takes up a descriptive approach to analyze the underpinnings of drug approval
process by both ‘the FDA and the EMA’. The methodology focuses on interpretation of the
secondary data. The data is both quantitative and qualitative. The study applies a reasoning to
comprehend the different unexamined aspects of oncology drug approval process, done by
EMA and FDA.
20

3.2 Research philosophy
As a research philosophy, positivism depicts the perspective that “factual” knowledge
is gained through the observation (more precisely, using the senses) that include assessments
and measurements (Morgenthau 2017). Researcher’s role in positivism studies is restricted to
a data collection and a systemic interpretation using the objective way. In studies with
positivism philosophy, the research results are quantifiable and observable. Positivism works
on quantifiable observations, leading to statistical analyses. As in philosophy, the positivism
theory is synchronized with the empiricism philosophy. The studies, words and the positivist
paradigm are dependent on the facts and it considers world from an external perspective and
from objective ideas as well. Particularly, positivism is based on the aspects of science which
are:- a. Science is deterministic. X can cause Y under the certain circumstances. It works on
the principle of cause and the effect relationship. b. Science is mechanistic - the researchers
develop the hypotheses that can be proved or even disproved, based on application of definite
research methods. c. Science uses method – The application of the methodology comprises
selection of the sample, the measurements and the analysis – finally reaching the conclusions
about proposed hypotheses. d. Science deals with empiricism – Science, in fact, can deal
with things that can be only measured or observed (Pierre 2016).
Interpretivism is a research that comprises of researchers who interpret the elements
of study and interpretivism assimilates human interest in the setting (Sullivan 2016).
Therefore, an interpretive research assume that reality’s access is only through the social
constructions like language, shared meanings, consciousness, imagination and the
instruments. A progress in interpretivist philosophy depends on positivism critique in the
social sciences. Accordingly, the research philosophy highlights qualitative analysis over any
quantitative analysis.
21
As a research philosophy, positivism depicts the perspective that “factual” knowledge
is gained through the observation (more precisely, using the senses) that include assessments
and measurements (Morgenthau 2017). Researcher’s role in positivism studies is restricted to
a data collection and a systemic interpretation using the objective way. In studies with
positivism philosophy, the research results are quantifiable and observable. Positivism works
on quantifiable observations, leading to statistical analyses. As in philosophy, the positivism
theory is synchronized with the empiricism philosophy. The studies, words and the positivist
paradigm are dependent on the facts and it considers world from an external perspective and
from objective ideas as well. Particularly, positivism is based on the aspects of science which
are:- a. Science is deterministic. X can cause Y under the certain circumstances. It works on
the principle of cause and the effect relationship. b. Science is mechanistic - the researchers
develop the hypotheses that can be proved or even disproved, based on application of definite
research methods. c. Science uses method – The application of the methodology comprises
selection of the sample, the measurements and the analysis – finally reaching the conclusions
about proposed hypotheses. d. Science deals with empiricism – Science, in fact, can deal
with things that can be only measured or observed (Pierre 2016).
Interpretivism is a research that comprises of researchers who interpret the elements
of study and interpretivism assimilates human interest in the setting (Sullivan 2016).
Therefore, an interpretive research assume that reality’s access is only through the social
constructions like language, shared meanings, consciousness, imagination and the
instruments. A progress in interpretivist philosophy depends on positivism critique in the
social sciences. Accordingly, the research philosophy highlights qualitative analysis over any
quantitative analysis.
21
Secure Best Marks with AI Grader
Need help grading? Try our AI Grader for instant feedback on your assignments.

Realism research philosophy is of the third type that depends on freedom from the
idea of reality from the human mind. Develop in knowledge is the core concept of this
philosophy. Realism is of two types – critical and direct. Direct realism – is to conceive
ideas as we see things and being bound to it. In different words, this direct realism depicts the
world through individual sensations (Rysiew 2017). Critical realism argues that humans
only experience parts, images and sensations of true world (Hartwig 2015).
This research philosophy used here, is ‘interpretivism’ as it interprets ‘secondary data
of studies’ that has already explored the differences and similarities between the ‘FDA’ and
the ‘EMA’. The study explores and discusses the findings from different perspectives in order
to elucidate the implications in a more vivid manner.
3.3 Research approach
Inductive approach also called inductive reasoning begins with observations and the
theories are research process based on observations (McAbee et al; 2017). Inductive research
comprises of the ‘observation patterns and their explanations’. The inductive approach cannot
be used at the beginning of any research.
Researchers using a deductive approach that starts with ‘social theory’ and then, the
researchers test the implications for data. They move from ‘general level to a very specific
level’ of data analysis. The deductive approach used in research is associated with scientific
investigation (Alfred 2017). Researcher studies the ‘already performed researches’, read and
comprehend about ‘previous phenomenon researches’ that she or he has been studying. The
researchers ‘test’ or scrutinize the hypotheses that emerge from scientific theories. Before an
investigation can happen, the basic approach is generally decided beforeand.
Abductive reasoning is used in an ‘abductive approach’ that address ‘the shortcomings’
associated with ‘inductive and deductive approaches’ (Dunne 2016). Particularly, the
22
idea of reality from the human mind. Develop in knowledge is the core concept of this
philosophy. Realism is of two types – critical and direct. Direct realism – is to conceive
ideas as we see things and being bound to it. In different words, this direct realism depicts the
world through individual sensations (Rysiew 2017). Critical realism argues that humans
only experience parts, images and sensations of true world (Hartwig 2015).
This research philosophy used here, is ‘interpretivism’ as it interprets ‘secondary data
of studies’ that has already explored the differences and similarities between the ‘FDA’ and
the ‘EMA’. The study explores and discusses the findings from different perspectives in order
to elucidate the implications in a more vivid manner.
3.3 Research approach
Inductive approach also called inductive reasoning begins with observations and the
theories are research process based on observations (McAbee et al; 2017). Inductive research
comprises of the ‘observation patterns and their explanations’. The inductive approach cannot
be used at the beginning of any research.
Researchers using a deductive approach that starts with ‘social theory’ and then, the
researchers test the implications for data. They move from ‘general level to a very specific
level’ of data analysis. The deductive approach used in research is associated with scientific
investigation (Alfred 2017). Researcher studies the ‘already performed researches’, read and
comprehend about ‘previous phenomenon researches’ that she or he has been studying. The
researchers ‘test’ or scrutinize the hypotheses that emerge from scientific theories. Before an
investigation can happen, the basic approach is generally decided beforeand.
Abductive reasoning is used in an ‘abductive approach’ that address ‘the shortcomings’
associated with ‘inductive and deductive approaches’ (Dunne 2016). Particularly, the
22

deductive reasoning have often criticized because it entends a ‘lack of clarity’ in analysis of
‘formulating hypotheses’. Abductive reasoning delivers a ‘decision’ based on a ‘very likely
inference’ derived from a ‘set of given observations’.
Many previous studies have already explained the various aspects of drug approval
process by EMA and FDA. This study uses an abductive approach to analyse the ‘logical
gaps’ left by previous researches which attempted to analyse the same problems associated
with drug approval.
3.4 Design
Exploratory research explores the research questions and do not provide conclusive and
final solutions to the existing problems (Claessen et al; 2016). This research type aid to
comprehend a study problem that is still elusive. The research explores the problem’s nature;
an exploratory research provides a conclusive evidence and helps us to develop a finer
understanding of research problem. When the exploratory research is conducted, the
researcher should be and is expected to shift directions, and change perspectives as the new
data and the new insights are revealed.
The descriptive research denotes the research questions, research design and the data
analysis that can be conducted in the topic (Colorafi and Evans 2016). The observational
research method assures that no variables are affected in its capacity. In quantitative research
– a descriptive research is quantitative in nature that attempts to gain quantifiable evidence
that is to be used during statistical analysis of population sample. Regarding ‘uncontrolled
variables’ - in the ‘descriptive researches’, no variables are affected in any manner. This
applies observational methods to the research in a particular way. The variables’ nature or the
progressive behavior is not in researcher’s hands. In cross-sectional studies – the descriptive
research deals with different sections that belong to a same group. The data that is collected
23
‘formulating hypotheses’. Abductive reasoning delivers a ‘decision’ based on a ‘very likely
inference’ derived from a ‘set of given observations’.
Many previous studies have already explained the various aspects of drug approval
process by EMA and FDA. This study uses an abductive approach to analyse the ‘logical
gaps’ left by previous researches which attempted to analyse the same problems associated
with drug approval.
3.4 Design
Exploratory research explores the research questions and do not provide conclusive and
final solutions to the existing problems (Claessen et al; 2016). This research type aid to
comprehend a study problem that is still elusive. The research explores the problem’s nature;
an exploratory research provides a conclusive evidence and helps us to develop a finer
understanding of research problem. When the exploratory research is conducted, the
researcher should be and is expected to shift directions, and change perspectives as the new
data and the new insights are revealed.
The descriptive research denotes the research questions, research design and the data
analysis that can be conducted in the topic (Colorafi and Evans 2016). The observational
research method assures that no variables are affected in its capacity. In quantitative research
– a descriptive research is quantitative in nature that attempts to gain quantifiable evidence
that is to be used during statistical analysis of population sample. Regarding ‘uncontrolled
variables’ - in the ‘descriptive researches’, no variables are affected in any manner. This
applies observational methods to the research in a particular way. The variables’ nature or the
progressive behavior is not in researcher’s hands. In cross-sectional studies – the descriptive
research deals with different sections that belong to a same group. The data that is collected
23

and then analyzed using various research techniques. The type of data collected or to be
collected determines the type of research approach.
In explanatory Research - The aim of an explanatory research is to increase the
researcher’s comprehension about a certain subject (Abderma et al; 2018). It does not make
any conclusive which is because it lacks the statistical perspective, but it makes the
researcher determine how and why things happen. Secondary sources that are published in
literature help in collection of data for a precision in explanatory research type. An extra care
should be taken in order to select the spectrum of a source that provide a wide and a well-
balanced comprehension of subject.
This study uses a descriptive approach to explain the implicit aspects of oncology
drug approval process by the ‘EMA’ and the ‘FDA’. Starting from the drug designing to
approval process to distribution to the markets – the entire process is analyzed reflectively
and critically in accordance with the laws, regulatory bodies, administrative underpinnings
prevalent in United States and Europe.
3.5 Data collection method
The data collection method used in this study is a ‘systemic secondary data
collection’. The secondary data can be quantitative and qualitative. ‘Secondary qualitative
data’ can be journals while ‘quantitative data’ can be obtained through a survey, journals and
the statistics. There is secondary data already available and this saves lot of time as compared
to the collection of primary data. In addition, secondary data can be obtained at a lesser
expense than primary data. Although, as data collected from various sources lacks a
specificity to the researcher’s needs and that is why, a more arduous data analysis has to be
undertaken in order to facilitate the same.
24
collected determines the type of research approach.
In explanatory Research - The aim of an explanatory research is to increase the
researcher’s comprehension about a certain subject (Abderma et al; 2018). It does not make
any conclusive which is because it lacks the statistical perspective, but it makes the
researcher determine how and why things happen. Secondary sources that are published in
literature help in collection of data for a precision in explanatory research type. An extra care
should be taken in order to select the spectrum of a source that provide a wide and a well-
balanced comprehension of subject.
This study uses a descriptive approach to explain the implicit aspects of oncology
drug approval process by the ‘EMA’ and the ‘FDA’. Starting from the drug designing to
approval process to distribution to the markets – the entire process is analyzed reflectively
and critically in accordance with the laws, regulatory bodies, administrative underpinnings
prevalent in United States and Europe.
3.5 Data collection method
The data collection method used in this study is a ‘systemic secondary data
collection’. The secondary data can be quantitative and qualitative. ‘Secondary qualitative
data’ can be journals while ‘quantitative data’ can be obtained through a survey, journals and
the statistics. There is secondary data already available and this saves lot of time as compared
to the collection of primary data. In addition, secondary data can be obtained at a lesser
expense than primary data. Although, as data collected from various sources lacks a
specificity to the researcher’s needs and that is why, a more arduous data analysis has to be
undertaken in order to facilitate the same.
24
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

A secondary data collection is used in order to understand the guidelines of FDA and
EMA and in order to bridge the gaps left by previous researches done on similar topics.
3.6 Data analysis
A ‘mixed-method data analysis’ is used in this study. Mixed methods research is a
methodology for conducting research that includes gathering, scrutinizing and assimilating
the quantitative (surveys, experiments) and qualitative (interviews, focus groups) research.
There are five underlying principles to a social research. Firstly, ‘social science’ is important
to a ‘democratic society’ and must be comprehensive of various interests, funders, values,
methods and aspects. Secondly, all the social sciences must respect the autonomy, privacy,
diversity, values and self-respect of the individuals, the groups and the communities. Thirdly,
All social sciences must be conducted with integrity through, recruiting the most suitable
methods to serve the research purpose. Fourthly, all social scientists must act in regards with
social responsibilities while conducting and distributing the research resources to the
participants. Fifthly, all social sciences must aim to increase benefit and reduce harm. This
mixed method to research, provides an integration and clarity to research problem than by
incorporating te best of two methods (qualitative and quantitative). By mixing both
quantitative and qualitative research and data, the researcher gains in breadth and depth of
understanding and corroboration, while offsetting the weaknesses inherent to using each
approach by itself.
3.7Summary
The study uses interpretivism as a research philosophy. Interpretivism would allow
the researchers to gain an insight into the social and cultural impacts of delayed and deficient
drug approval process. The philosophy also allowed the researchers to interpret FDA and
EMA’s impact of overall public health service in United States and Europe respectively. In
research approach, an abductive approach has been into order to close the shortcomings of
25
EMA and in order to bridge the gaps left by previous researches done on similar topics.
3.6 Data analysis
A ‘mixed-method data analysis’ is used in this study. Mixed methods research is a
methodology for conducting research that includes gathering, scrutinizing and assimilating
the quantitative (surveys, experiments) and qualitative (interviews, focus groups) research.
There are five underlying principles to a social research. Firstly, ‘social science’ is important
to a ‘democratic society’ and must be comprehensive of various interests, funders, values,
methods and aspects. Secondly, all the social sciences must respect the autonomy, privacy,
diversity, values and self-respect of the individuals, the groups and the communities. Thirdly,
All social sciences must be conducted with integrity through, recruiting the most suitable
methods to serve the research purpose. Fourthly, all social scientists must act in regards with
social responsibilities while conducting and distributing the research resources to the
participants. Fifthly, all social sciences must aim to increase benefit and reduce harm. This
mixed method to research, provides an integration and clarity to research problem than by
incorporating te best of two methods (qualitative and quantitative). By mixing both
quantitative and qualitative research and data, the researcher gains in breadth and depth of
understanding and corroboration, while offsetting the weaknesses inherent to using each
approach by itself.
3.7Summary
The study uses interpretivism as a research philosophy. Interpretivism would allow
the researchers to gain an insight into the social and cultural impacts of delayed and deficient
drug approval process. The philosophy also allowed the researchers to interpret FDA and
EMA’s impact of overall public health service in United States and Europe respectively. In
research approach, an abductive approach has been into order to close the shortcomings of
25

secondary research findings. A descriptive research approach was chosen because the study
aims to broadly describe the defects, present in the cancer drug approval process of FDA and
EMA. To serve the purpose, a secondary data collection has been undertaken and a mixed
data analysis of the quantitative and qualitative data has been done.
Chapter 4 Results and Discussion
4.1Drug Approval in the United States
The United States, by a significant margin, have considerably very stringent standards
in approval of new drugs (Van Norman 2016). The drug approval process and standards in
the United States are considered by others to be very complex and very demanding at the
same time. It begins with an investigational New Drug Application, which is then filed to the
FDA and it is thereafter, they send a set of instructions of human clinical trials (Bobo et al;
2016) with the reports of the Preclinical trials (Bailey et al; 2014). Pre - IND meetings are
pertinent to the FDA while discussing various issues. The animal research design that is
needed to lend a support to various clinical studies – is important as well. Intended protocol
(Ciociola et al; 2014) are needed for conductance of different clinical trials. Deciding a
chemical composition, the manufacturing, and the control of investigational drug is vital to
the sustenance of whole pharmaceutical approval process. The organization works very
closely with the sponsor in order to organize an animal research, to gather data, to design
clinical protocols dependent on the FDA suggestions.
New Drug Application or NDA – when the clinical studies are confirmed for safety
and efficiency of new drug and it poses no unreasonable threats and dangers to the patients
(Mullard 2015) - the next process begins. It is then the manufacturer sends a New Drug
Application, which is an actual request to sell and manufacture drugs in the United States.
Abbreviated New Drug Application or ANDA is an application made for generic drugs
26
aims to broadly describe the defects, present in the cancer drug approval process of FDA and
EMA. To serve the purpose, a secondary data collection has been undertaken and a mixed
data analysis of the quantitative and qualitative data has been done.
Chapter 4 Results and Discussion
4.1Drug Approval in the United States
The United States, by a significant margin, have considerably very stringent standards
in approval of new drugs (Van Norman 2016). The drug approval process and standards in
the United States are considered by others to be very complex and very demanding at the
same time. It begins with an investigational New Drug Application, which is then filed to the
FDA and it is thereafter, they send a set of instructions of human clinical trials (Bobo et al;
2016) with the reports of the Preclinical trials (Bailey et al; 2014). Pre - IND meetings are
pertinent to the FDA while discussing various issues. The animal research design that is
needed to lend a support to various clinical studies – is important as well. Intended protocol
(Ciociola et al; 2014) are needed for conductance of different clinical trials. Deciding a
chemical composition, the manufacturing, and the control of investigational drug is vital to
the sustenance of whole pharmaceutical approval process. The organization works very
closely with the sponsor in order to organize an animal research, to gather data, to design
clinical protocols dependent on the FDA suggestions.
New Drug Application or NDA – when the clinical studies are confirmed for safety
and efficiency of new drug and it poses no unreasonable threats and dangers to the patients
(Mullard 2015) - the next process begins. It is then the manufacturer sends a New Drug
Application, which is an actual request to sell and manufacture drugs in the United States.
Abbreviated New Drug Application or ANDA is an application made for generic drugs
26
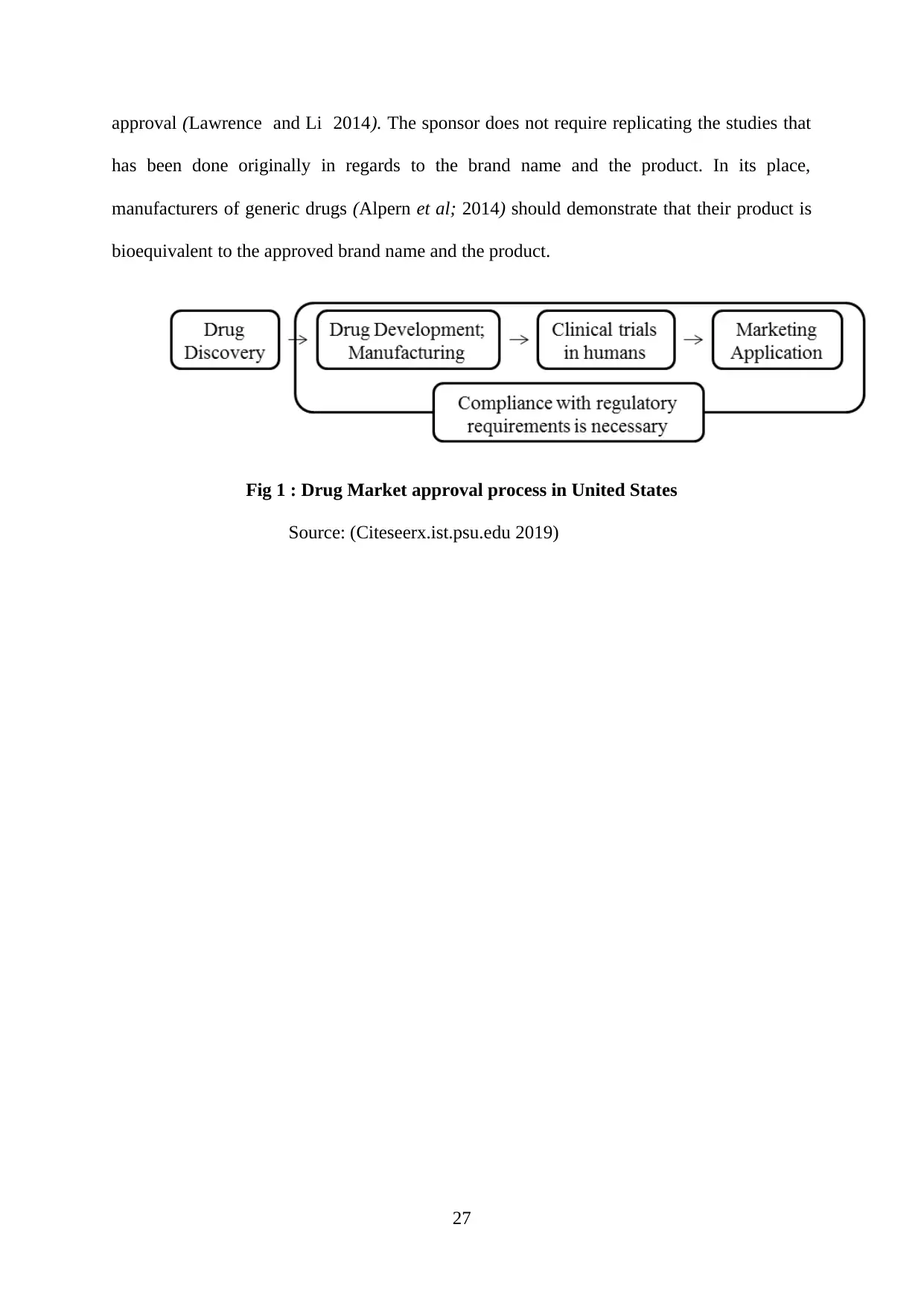
approval (Lawrence and Li 2014). The sponsor does not require replicating the studies that
has been done originally in regards to the brand name and the product. In its place,
manufacturers of generic drugs (Alpern et al; 2014) should demonstrate that their product is
bioequivalent to the approved brand name and the product.
Fig 1 : Drug Market approval process in United States
Source: (Citeseerx.ist.psu.edu 2019)
27
has been done originally in regards to the brand name and the product. In its place,
manufacturers of generic drugs (Alpern et al; 2014) should demonstrate that their product is
bioequivalent to the approved brand name and the product.
Fig 1 : Drug Market approval process in United States
Source: (Citeseerx.ist.psu.edu 2019)
27
Secure Best Marks with AI Grader
Need help grading? Try our AI Grader for instant feedback on your assignments.

Figure 2: New Drug Application
Source: (Citeseerx.ist.psu.edu 2019)
28
Source: (Citeseerx.ist.psu.edu 2019)
28

Figure 3: Generic Drugs Approval
Source: (Citeseerx.ist.psu.edu 2019)
4.2Drug Approval in Europe
Similar to the United States requirements – there are 2 regulatory steps that must be
adhered to, prior to drug approval for marketing in the European Union. These 2 steps are
known as ‘clinical trial application’, ‘marketing authorization application’ (Elsäßer et al;
2014). In all, there were twenty-seven member states within the European Union (as in
August of 2007). These clinical Trial Applications (Bonini et al; 2014) are state level
approved whereas the marketing authorization applications can be approved at member state
and at centralized levels (Hoekman et al; 2015).This happens in a centralized procedure
29
Source: (Citeseerx.ist.psu.edu 2019)
4.2Drug Approval in Europe
Similar to the United States requirements – there are 2 regulatory steps that must be
adhered to, prior to drug approval for marketing in the European Union. These 2 steps are
known as ‘clinical trial application’, ‘marketing authorization application’ (Elsäßer et al;
2014). In all, there were twenty-seven member states within the European Union (as in
August of 2007). These clinical Trial Applications (Bonini et al; 2014) are state level
approved whereas the marketing authorization applications can be approved at member state
and at centralized levels (Hoekman et al; 2015).This happens in a centralized procedure
29

(Alqahtani et al; 2015). in which the applicants receive a marketing authorization that is
validated through EU. Single authorization results are valid in Norway, EU, Liechtenstein
and Iceland.
The EMA has a timeline of 210 days and it submits the approved requests to the
European Commission in order to get a final approval. Centralized process is important for
medicines that are derived from biotechnology processes like genetic engineering. The
medicines are intended for cancer interventions, diabetes, HIV/Aids, neurodegenerative
disorders, the autoimmune diseases, also immune dysfunctions. The oncology medicines are
officially designated as 'orphan medicines' as they are used in rarest of situations. In the
mutual Recognition Procedure – the applicants are allowed to acquire marketing
authorization within concerned member states other than the Reference Member States. The
applicant then submits the same dossier to member states of EU that require authorization
regarding the information. The sooner the member state chooses to assess medicinal products
– it becomes RMS notifying their decision to different other states. The member States are
called "CMS" through those and to whom, these applications are submitted (Pacurariu et al;
2014). The RMS then sends a report to the other states based on their own findings (Vishal et
al; 2014). The generic industry is cardinal user in this type of the drug approval procedure.
The method or procedure can take up to timeline of three hundred and ninety days. The
Nationalized Procedure, on the other hand is the one that permit applicants to acquire a
marketing authorization within one member state only. In order to receive a national
marketing authorization, the application can be submitted to competent authority of Member
State (Leyens and Brand 2016). The new substances, in Centralized procedure, can gain
marketing authorization with this procedure. Timeline for national procedure is 210 Days.
Using the decentralized procedure (Garattini and Padula 2017), the companies can
apply for authorized duty for in one EU country. Based on the assessment report made by
30
validated through EU. Single authorization results are valid in Norway, EU, Liechtenstein
and Iceland.
The EMA has a timeline of 210 days and it submits the approved requests to the
European Commission in order to get a final approval. Centralized process is important for
medicines that are derived from biotechnology processes like genetic engineering. The
medicines are intended for cancer interventions, diabetes, HIV/Aids, neurodegenerative
disorders, the autoimmune diseases, also immune dysfunctions. The oncology medicines are
officially designated as 'orphan medicines' as they are used in rarest of situations. In the
mutual Recognition Procedure – the applicants are allowed to acquire marketing
authorization within concerned member states other than the Reference Member States. The
applicant then submits the same dossier to member states of EU that require authorization
regarding the information. The sooner the member state chooses to assess medicinal products
– it becomes RMS notifying their decision to different other states. The member States are
called "CMS" through those and to whom, these applications are submitted (Pacurariu et al;
2014). The RMS then sends a report to the other states based on their own findings (Vishal et
al; 2014). The generic industry is cardinal user in this type of the drug approval procedure.
The method or procedure can take up to timeline of three hundred and ninety days. The
Nationalized Procedure, on the other hand is the one that permit applicants to acquire a
marketing authorization within one member state only. In order to receive a national
marketing authorization, the application can be submitted to competent authority of Member
State (Leyens and Brand 2016). The new substances, in Centralized procedure, can gain
marketing authorization with this procedure. Timeline for national procedure is 210 Days.
Using the decentralized procedure (Garattini and Padula 2017), the companies can
apply for authorized duty for in one EU country. Based on the assessment report made by
30
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser
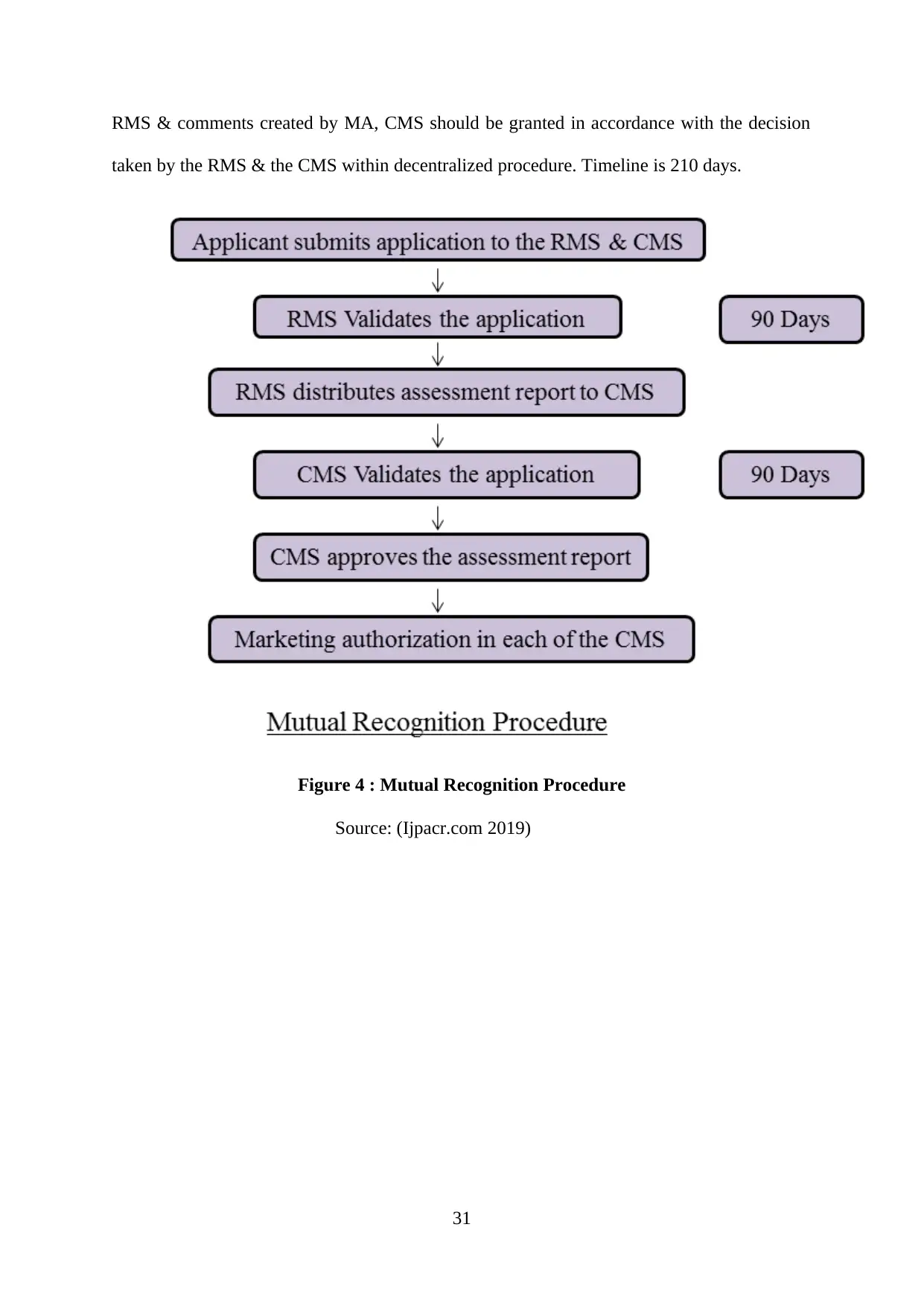
RMS & comments created by MA, CMS should be granted in accordance with the decision
taken by the RMS & the CMS within decentralized procedure. Timeline is 210 days.
Figure 4 : Mutual Recognition Procedure
Source: (Ijpacr.com 2019)
31
taken by the RMS & the CMS within decentralized procedure. Timeline is 210 days.
Figure 4 : Mutual Recognition Procedure
Source: (Ijpacr.com 2019)
31

Figure 5: Decentralised Procedure
Source: (Ijpacr.com 2019)
Here is a brief comparison that is shown as the procedures followed by regulatory
agencies. Figure 5 shows the comparative study done by Dossier while submitting a drug. In
the administrative section - administrative prerequisites like the application, the copies and its
numbers, the fees and the type of the presentation is required and is mentioned. The
important facets are – 1. ‘Finished Product Control’ (Krcevski-Skvarc et al; 2018)- where the
requirements are very clearly stated. 2. ‘Manufacturing and Control’ - requirements well
32
Source: (Ijpacr.com 2019)
Here is a brief comparison that is shown as the procedures followed by regulatory
agencies. Figure 5 shows the comparative study done by Dossier while submitting a drug. In
the administrative section - administrative prerequisites like the application, the copies and its
numbers, the fees and the type of the presentation is required and is mentioned. The
important facets are – 1. ‘Finished Product Control’ (Krcevski-Skvarc et al; 2018)- where the
requirements are very clearly stated. 2. ‘Manufacturing and Control’ - requirements well
32

stated. 3. Stability - requirements should be well stated. 4. Bioequivalence - Requirements
should be well stated. 5. ‘World Parma Market’ – should be taken into comparison.
From January of 1999 to May of 2014, the EMA has issued about ‘75’ approvals, that
includes about ‘44’ indications approved solely based on the uncontrolled trials. Of forty-four
approvals, ‘8’ of the treatment indications are RCT approved indications, while the remaining
‘36’ approvals were resultant of no RCT approved indications (Monami et al; 2014). Over
time-period, about 9 applications without the support of RCT results, the ‘EMA’ rejected
application and the oncology drug’s manufacturer have withdrawn from the same with a
negative recommendation. In identical time-period, the FDA produced about ‘74’ approvals.
This included about ‘60’ of the indications that are solely approved based out of the
uncontrolled studies. Of the sixty approvals, ‘12’ are extensions of the treatment indications
with the RCTs in approved indications. The rest ‘48’ were the products of randomized
control trials done in approved indications. There was only ‘1’ application, which was made
without any randomized controlled trial results that was disapproved.
De-duplicating the regional approvals, ‘74’ indications were approved in one period
by one agency performing uncontrolled trials. Out of these, ‘34’ approvals were from
hematological oncology, ‘15’ were from solid tumor oncology and ‘15’ were from rare
enzyme replacement therapies. Out of these areas, about 8 approvals were granted in
emergency medicine, poisoning and 2 were granted in general hematology. These approvals
were license extensions for the existent therapies in solid tumor oncology and hematological
oncology. Searching the treatments approved by the one agency based only on uncontrolled
study date, four of such approvals was used in comparison of the ‘FDA’ and the ‘EMA’
approval times and rates. The ‘EMA’ approved ‘4 treatments’ in 1999 that were approved
prior to the FDA with similar type of data package in same year.
33
should be well stated. 5. ‘World Parma Market’ – should be taken into comparison.
From January of 1999 to May of 2014, the EMA has issued about ‘75’ approvals, that
includes about ‘44’ indications approved solely based on the uncontrolled trials. Of forty-four
approvals, ‘8’ of the treatment indications are RCT approved indications, while the remaining
‘36’ approvals were resultant of no RCT approved indications (Monami et al; 2014). Over
time-period, about 9 applications without the support of RCT results, the ‘EMA’ rejected
application and the oncology drug’s manufacturer have withdrawn from the same with a
negative recommendation. In identical time-period, the FDA produced about ‘74’ approvals.
This included about ‘60’ of the indications that are solely approved based out of the
uncontrolled studies. Of the sixty approvals, ‘12’ are extensions of the treatment indications
with the RCTs in approved indications. The rest ‘48’ were the products of randomized
control trials done in approved indications. There was only ‘1’ application, which was made
without any randomized controlled trial results that was disapproved.
De-duplicating the regional approvals, ‘74’ indications were approved in one period
by one agency performing uncontrolled trials. Out of these, ‘34’ approvals were from
hematological oncology, ‘15’ were from solid tumor oncology and ‘15’ were from rare
enzyme replacement therapies. Out of these areas, about 8 approvals were granted in
emergency medicine, poisoning and 2 were granted in general hematology. These approvals
were license extensions for the existent therapies in solid tumor oncology and hematological
oncology. Searching the treatments approved by the one agency based only on uncontrolled
study date, four of such approvals was used in comparison of the ‘FDA’ and the ‘EMA’
approval times and rates. The ‘EMA’ approved ‘4 treatments’ in 1999 that were approved
prior to the FDA with similar type of data package in same year.
33
Secure Best Marks with AI Grader
Need help grading? Try our AI Grader for instant feedback on your assignments.

For applications done in both of the regions, forty-four of the applications were the
‘EMA’, the ‘FDA’ approved, without controlled results (including the 4 approvals made by
FDA prior to the year of 1999). Out of thirty-four applications approved in both of the
regions, the EMA approval was given a mean of about 13.4 months after (IQR 4.5–17.2
months, median 6.7). This delay is divided into two parts – at the first, companies that
submitted to EMA had a mean of about 7.4 months after (IQR 0.1–8 months, median 1.5) and
28 out of these 34 indications were submitted to the FDA at first. Secondly, the EMA took
6.3 months average prolonged time to complete the review and to approve the products (IQR
0.1–0.3 months, median 0.2). In equivalent approvals, the EMA’s review was faster in 3 out
of the 34 applications that leads to 30 out of 34 products distributed faster in the ‘USA
markets’. Additionally, the ‘EMA’ approving of fewer products and then delaying while
approving of the products, 5 treatments were approved by the ‘FDA’ using uncontrolled
studies but approval by the ‘EMA’ later on when the comparative results was present (an
approval delay of about 21.5 months mean/9 months of median).
The approvals done without the support of RCT evidence were found to be in
astonishingly high numbers and exceeded the expectations and its ‘mean value’ was nearly
equal to 5 indications every year as it was approved by both (or either) of the EMA and of the
FDA. The license extensions in existing products were only about nineteen percent of the
approvals that were demonstrated to be efficacious in the randomized control trials of
diseases concerned. Though, the approvals based just on uncontrolled data were of relatively
small numbers as compared to total approvals. The study finds out that the ‘EMA’ approves 3
of such cases per year and the ‘FDA’ approves about 4 per year. These are then subjected to
further scrutiny. The pathologic areas where the uncontrolled studies are applied in approval
process were of cardinal interest in oncology – 49 out of 74 indications that is sixty-six
percent were either solid tumor or hematological oncology. This corresponds with previous
34
‘EMA’, the ‘FDA’ approved, without controlled results (including the 4 approvals made by
FDA prior to the year of 1999). Out of thirty-four applications approved in both of the
regions, the EMA approval was given a mean of about 13.4 months after (IQR 4.5–17.2
months, median 6.7). This delay is divided into two parts – at the first, companies that
submitted to EMA had a mean of about 7.4 months after (IQR 0.1–8 months, median 1.5) and
28 out of these 34 indications were submitted to the FDA at first. Secondly, the EMA took
6.3 months average prolonged time to complete the review and to approve the products (IQR
0.1–0.3 months, median 0.2). In equivalent approvals, the EMA’s review was faster in 3 out
of the 34 applications that leads to 30 out of 34 products distributed faster in the ‘USA
markets’. Additionally, the ‘EMA’ approving of fewer products and then delaying while
approving of the products, 5 treatments were approved by the ‘FDA’ using uncontrolled
studies but approval by the ‘EMA’ later on when the comparative results was present (an
approval delay of about 21.5 months mean/9 months of median).
The approvals done without the support of RCT evidence were found to be in
astonishingly high numbers and exceeded the expectations and its ‘mean value’ was nearly
equal to 5 indications every year as it was approved by both (or either) of the EMA and of the
FDA. The license extensions in existing products were only about nineteen percent of the
approvals that were demonstrated to be efficacious in the randomized control trials of
diseases concerned. Though, the approvals based just on uncontrolled data were of relatively
small numbers as compared to total approvals. The study finds out that the ‘EMA’ approves 3
of such cases per year and the ‘FDA’ approves about 4 per year. These are then subjected to
further scrutiny. The pathologic areas where the uncontrolled studies are applied in approval
process were of cardinal interest in oncology – 49 out of 74 indications that is sixty-six
percent were either solid tumor or hematological oncology. This corresponds with previous
34

work that is regarding the drug licensing, which poses a lesser barrier to oncology medicine
approval. Of the nine applications presented to the ‘EMA’ which were not approved, 7 were
found to be in oncology, the ‘EMA’ then highlights the uncertainty regarding benefit to
treatment risk ratio.
While the unmet needs in oncology (as perceived) has been used as argument in use
of the uncontrolled studies. Additionally, it is feasible that ‘FDA’ does not approve the
treatments, which were approved by ‘EMA’. It is unlikely that it remains a very unavoidable
constraint of the study. The study finds out a substantial overlap in the decisions and it
reveals that FDA is more so over ready to approve the products (drugs) on basis of the
uncontrolled trials. The mentioned difference reflect on attitudes of regulators toward the risk
versus the unmet health and medical needs.
Other evidence reveal that 2 systems can produce decisions on different timescales,
while accessing similar studies. Difference in approval time has been observed in approval of
the tyrosine kinase inhibitor – thirteen oncology drugs and generic pharmaceuticals by 2
agencies. The difference is reasoned by the ‘FDA’s’ explicit use of ‘accelerated approvals’
with confirmation (Mullard 2015). Randomized control trials conducted subsequently was
compared to the lesser use of correspondent the ‘EMA’ process that is conditional approval.
The results regarding the approval dates revealed that the patients under the framework of the
‘EU’ has to wait very long for the novel treatments. Submitting delays between these two
agencies is comprehensible and provided – that the kind of staff involved in trials can be
required to formulate and scrutinize submissions for two of the agencies. The study finds out
that the companies prioritizes the FDA submission over the ‘EMA’. Initiatives undertaken to
harmonize processes (such as transatlantic trade) can be helpful, provided there are no
negative externalities. Problem with prolonged approval periods for the ‘EMA’ is very
complex. In Europe, the longer duration can be due to the lack of rapid organizational
35
approval. Of the nine applications presented to the ‘EMA’ which were not approved, 7 were
found to be in oncology, the ‘EMA’ then highlights the uncertainty regarding benefit to
treatment risk ratio.
While the unmet needs in oncology (as perceived) has been used as argument in use
of the uncontrolled studies. Additionally, it is feasible that ‘FDA’ does not approve the
treatments, which were approved by ‘EMA’. It is unlikely that it remains a very unavoidable
constraint of the study. The study finds out a substantial overlap in the decisions and it
reveals that FDA is more so over ready to approve the products (drugs) on basis of the
uncontrolled trials. The mentioned difference reflect on attitudes of regulators toward the risk
versus the unmet health and medical needs.
Other evidence reveal that 2 systems can produce decisions on different timescales,
while accessing similar studies. Difference in approval time has been observed in approval of
the tyrosine kinase inhibitor – thirteen oncology drugs and generic pharmaceuticals by 2
agencies. The difference is reasoned by the ‘FDA’s’ explicit use of ‘accelerated approvals’
with confirmation (Mullard 2015). Randomized control trials conducted subsequently was
compared to the lesser use of correspondent the ‘EMA’ process that is conditional approval.
The results regarding the approval dates revealed that the patients under the framework of the
‘EU’ has to wait very long for the novel treatments. Submitting delays between these two
agencies is comprehensible and provided – that the kind of staff involved in trials can be
required to formulate and scrutinize submissions for two of the agencies. The study finds out
that the companies prioritizes the FDA submission over the ‘EMA’. Initiatives undertaken to
harmonize processes (such as transatlantic trade) can be helpful, provided there are no
negative externalities. Problem with prolonged approval periods for the ‘EMA’ is very
complex. In Europe, the longer duration can be due to the lack of rapid organizational
35

decisions making as it includes all the EU member states (like Liechtenstein, Iceland and
Norway). Another cardinal factor in higher non-approval degree by the ‘EMA’ and the taking
of longer review period is – the chance for discussion about the applications content along
with the resolution of issues before an official dossier is submitted. These companies engage
with the ‘FDA’ after every phase of clinical development and after New Drug Application
(NDA) application (Ciociola et al; 2014). In Europe though, the drug manufacturing
companies does not essentially interact with EMA while the drug development is going on.
These companies do take Scientific Advice which is prior to submitting – that is only
fourteen out of the thirty-four products that is approved by both the agencies.
The study reveals the usually stated factors that affect the evidence availability that is
the rarity of situation and absence of the accepted treatments. The suitability of these
arguments vary and the comparative trials in rarest of conditions are perhaps challenging; but
not consistent. As example, the Canakinumab drug used for a Cryopyrin-associated periodic
syndrome (Kuemmerle-Deschner et al; 2017) has prevalence of less than 1 out of 100 000, as
reported from RCT of thirty-one patients. The study highlights the fact that are no specific
standardized plan of care, particularly for the terminal disease and in some cases – this was
used as a justification for usage of uncontrolled study. Under identical conditions, though
(Pixantrone drug for the aggression of relapsed non-Hodgkins lymphoma) - the manufacturer
ran a trial against physicians’ treatment choice. Even under the identical conditions, the
evidence base provided by companies was varied.
Even in situations, where the placebo Randomized control trials were very unviable -
the serious situations without the licensed treatments and a well-established outcome
poisoning is the obvious exam - the new treatments are often put against standardized care or
as complement to the existing therapy. These randomized controlled study designs is
extensively used in evaluation of the novelistic treatments for oncology and although, without
36
Norway). Another cardinal factor in higher non-approval degree by the ‘EMA’ and the taking
of longer review period is – the chance for discussion about the applications content along
with the resolution of issues before an official dossier is submitted. These companies engage
with the ‘FDA’ after every phase of clinical development and after New Drug Application
(NDA) application (Ciociola et al; 2014). In Europe though, the drug manufacturing
companies does not essentially interact with EMA while the drug development is going on.
These companies do take Scientific Advice which is prior to submitting – that is only
fourteen out of the thirty-four products that is approved by both the agencies.
The study reveals the usually stated factors that affect the evidence availability that is
the rarity of situation and absence of the accepted treatments. The suitability of these
arguments vary and the comparative trials in rarest of conditions are perhaps challenging; but
not consistent. As example, the Canakinumab drug used for a Cryopyrin-associated periodic
syndrome (Kuemmerle-Deschner et al; 2017) has prevalence of less than 1 out of 100 000, as
reported from RCT of thirty-one patients. The study highlights the fact that are no specific
standardized plan of care, particularly for the terminal disease and in some cases – this was
used as a justification for usage of uncontrolled study. Under identical conditions, though
(Pixantrone drug for the aggression of relapsed non-Hodgkins lymphoma) - the manufacturer
ran a trial against physicians’ treatment choice. Even under the identical conditions, the
evidence base provided by companies was varied.
Even in situations, where the placebo Randomized control trials were very unviable -
the serious situations without the licensed treatments and a well-established outcome
poisoning is the obvious exam - the new treatments are often put against standardized care or
as complement to the existing therapy. These randomized controlled study designs is
extensively used in evaluation of the novelistic treatments for oncology and although, without
36
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

the challenges, might be used to measure how new interventions are performed. There has
been various researches, which test the level of RCT’s effectiveness, at different levels. In the
treatments that has been identified, the persuasiveness of the evidence is to be referred.
As for example, a drug named imatinib is used to treat chronic myeloid leukemia
showed about 49% response rate for the major cytogenetic effect and thirty percent of total
cytogenetic response (Prasad and Mailankody 2014) was reported to be in five hundred and
thirty two patients while omacetaxine (in chronic myeloid leukemia) depicted a 18.4 percent
of cytogenetic responses. There was about 7.9% of cytogenetic response in all of seventy-six
patients. The fact is highlighted that approval without RCT, although unsafe, do not influence
the quality ‘data package’ in every manner. It is apparent that the most products are assessed
by the organizations like the ‘National Institute for Health and Care Excellence’ (NICE) that
play a critical role in estimation of the outcomes differences present between new treatments
with standardized care; the efficacy demonstration is not sufficient, alone. If the studies are
used in marketing and authorization of the products, it is not completely evident for
accessibility into the target markets (that is to convince the buyers that products is worth the
money). Then in various cases, application of uncontrolled trials is increasingly tough to
validate, that proofs are not required to qualify a medicine, which can be used in oncologic
treatment of the patients that critical levels. The scientific research that are based on
treatments conducts, are also approved without the randomized control trial results (that is
limited to the cancer drugs validated in United States of America). This reflects RCT’s
availability of evidence that does not influence the length of market stay and this leads to lack
of evidence or efficacy regarding safety of the drug without any randomized control trial
evidence available.
The researchers of this study found the fact concerning that the lack of confirmatory
evidence in the operation of the approval process, can lead to higher morbidity and mortality
37
been various researches, which test the level of RCT’s effectiveness, at different levels. In the
treatments that has been identified, the persuasiveness of the evidence is to be referred.
As for example, a drug named imatinib is used to treat chronic myeloid leukemia
showed about 49% response rate for the major cytogenetic effect and thirty percent of total
cytogenetic response (Prasad and Mailankody 2014) was reported to be in five hundred and
thirty two patients while omacetaxine (in chronic myeloid leukemia) depicted a 18.4 percent
of cytogenetic responses. There was about 7.9% of cytogenetic response in all of seventy-six
patients. The fact is highlighted that approval without RCT, although unsafe, do not influence
the quality ‘data package’ in every manner. It is apparent that the most products are assessed
by the organizations like the ‘National Institute for Health and Care Excellence’ (NICE) that
play a critical role in estimation of the outcomes differences present between new treatments
with standardized care; the efficacy demonstration is not sufficient, alone. If the studies are
used in marketing and authorization of the products, it is not completely evident for
accessibility into the target markets (that is to convince the buyers that products is worth the
money). Then in various cases, application of uncontrolled trials is increasingly tough to
validate, that proofs are not required to qualify a medicine, which can be used in oncologic
treatment of the patients that critical levels. The scientific research that are based on
treatments conducts, are also approved without the randomized control trial results (that is
limited to the cancer drugs validated in United States of America). This reflects RCT’s
availability of evidence that does not influence the length of market stay and this leads to lack
of evidence or efficacy regarding safety of the drug without any randomized control trial
evidence available.
The researchers of this study found the fact concerning that the lack of confirmatory
evidence in the operation of the approval process, can lead to higher morbidity and mortality
37

rates. However, any reassessment undertaken with the newly licensed drugs can elevate the
importance of initiatives like adaptive licensing. Limited information that is collected (on 5
drugs approved with uncontrolled data by FDA, USA and then by EMA after availability of
the Randomized control trial results indicate that regulatory assessments make thorough
decisions even in RCT’s absence but all these test designs were limited by small sample size.
There was a very specific concern found by this research, circulating around a sound use of
‘historical controls’ in the designs to compare the trial outcomes. The previous researches has
highlighted the exaggeration in the delivered ‘treatment effect’, with these historical controls
having performed worse than its equivalent arms within clinical studies.
The study inspects that the drug regulatory officials who has attempted to solve the
inconsistencies that influences development within the processes existent in pharmaceuticals
sectors across various countries. In the year of 1979, the ‘FDA’ started the ‘International
Affairs Staff’ in order to liaise along with the international regulatory services and with the
agencies providing them. A year later, the World Health Organization (WHO) and the FDA
hosted the first International Conference of Drug Regulatory Authorities. In the year of 1990,
Europe, United States and also Japan shook hands on ‘International Conference on the
Harmonization or ICH of Technical Requirements for Registration of Pharmaceuticals for
Human Use’ in order to safeguard a opportune introduction to new medical devices and
products, to mutual acceptance ways of research data, to an improved protection and
futuristic development of public health. The ICH has guidelines to standardize for the clinical
practices and the technical requirements in formulation of human drugs, including
establishment and the management in quality control setups and laboratories along with
prompt dissemination in terms of new information and adversative drug effects.
In recent times, both of the organization – the ‘FDA’ and the ‘EMA’ promoted
exchange in scientific knowledge across these agencies. In the year of 2005, the EMA, the
38
importance of initiatives like adaptive licensing. Limited information that is collected (on 5
drugs approved with uncontrolled data by FDA, USA and then by EMA after availability of
the Randomized control trial results indicate that regulatory assessments make thorough
decisions even in RCT’s absence but all these test designs were limited by small sample size.
There was a very specific concern found by this research, circulating around a sound use of
‘historical controls’ in the designs to compare the trial outcomes. The previous researches has
highlighted the exaggeration in the delivered ‘treatment effect’, with these historical controls
having performed worse than its equivalent arms within clinical studies.
The study inspects that the drug regulatory officials who has attempted to solve the
inconsistencies that influences development within the processes existent in pharmaceuticals
sectors across various countries. In the year of 1979, the ‘FDA’ started the ‘International
Affairs Staff’ in order to liaise along with the international regulatory services and with the
agencies providing them. A year later, the World Health Organization (WHO) and the FDA
hosted the first International Conference of Drug Regulatory Authorities. In the year of 1990,
Europe, United States and also Japan shook hands on ‘International Conference on the
Harmonization or ICH of Technical Requirements for Registration of Pharmaceuticals for
Human Use’ in order to safeguard a opportune introduction to new medical devices and
products, to mutual acceptance ways of research data, to an improved protection and
futuristic development of public health. The ICH has guidelines to standardize for the clinical
practices and the technical requirements in formulation of human drugs, including
establishment and the management in quality control setups and laboratories along with
prompt dissemination in terms of new information and adversative drug effects.
In recent times, both of the organization – the ‘FDA’ and the ‘EMA’ promoted
exchange in scientific knowledge across these agencies. In the year of 2005, the EMA, the
38

‘FDA’ and the ‘European Commission’ finalized 7 initiatives to share any information
comprising of accelerated approval, the design issues and the post-marketing obligations. 2
agencies communicated through monthly teleconferences in order to discuss the pending
reviews, regulatory decisions, and the requests for cessation of the clinical trials and if there
is any important changes amongst the statistical analyses results. Additionally, through
Transatlantic Economic Council (Cremona 2015) - the US and the EU seeks to deepen the
cooperation amidst these regulatory authorities so that there can be a reduction of
international duplication in the inspections (Bertrand et al; 2018). In the year 2011, these
agencies begun a pilot program in order to collaboratively measure the quality-by-design
components to review the science undertaken and the risks of the pharmaceutical
development in order to safeguard the product quality. They organized collaborative meetings
about pharmaco-vigilance (Nikfarjam et al; 2015) about concentrating on the biosimilars, the
oncological therapeutics, the orphan medicines and the pediatric drugs. Both the agencies
vowed to continue cross relationship activities in order to streamline and to synchronize the
transatlantic regulation.
In the present situation, the regulatory differences between the ‘US’ and the ‘EU’ can
hinder the expansion of the pharmaceutical markets. Functional and structural differences
between the EMA and the FDA exists and this leads to adequate planning in navigation of the
requirements that manufacture and then commercialize the medical products on an
international form (Lee 2014). Furthermore, collaborative engagements done between the
government agencies, the health advocates and the industry leaders who were pushed for an
all-inclusive process putting the pharmaceuticals industry in public context, more and more
securely and quickly. Futuristic regulatory standardization in between the agencies is vital to
minimize redundancy and also accelerate review process in workload with regards to
stakeholders’ benefits.
39
comprising of accelerated approval, the design issues and the post-marketing obligations. 2
agencies communicated through monthly teleconferences in order to discuss the pending
reviews, regulatory decisions, and the requests for cessation of the clinical trials and if there
is any important changes amongst the statistical analyses results. Additionally, through
Transatlantic Economic Council (Cremona 2015) - the US and the EU seeks to deepen the
cooperation amidst these regulatory authorities so that there can be a reduction of
international duplication in the inspections (Bertrand et al; 2018). In the year 2011, these
agencies begun a pilot program in order to collaboratively measure the quality-by-design
components to review the science undertaken and the risks of the pharmaceutical
development in order to safeguard the product quality. They organized collaborative meetings
about pharmaco-vigilance (Nikfarjam et al; 2015) about concentrating on the biosimilars, the
oncological therapeutics, the orphan medicines and the pediatric drugs. Both the agencies
vowed to continue cross relationship activities in order to streamline and to synchronize the
transatlantic regulation.
In the present situation, the regulatory differences between the ‘US’ and the ‘EU’ can
hinder the expansion of the pharmaceutical markets. Functional and structural differences
between the EMA and the FDA exists and this leads to adequate planning in navigation of the
requirements that manufacture and then commercialize the medical products on an
international form (Lee 2014). Furthermore, collaborative engagements done between the
government agencies, the health advocates and the industry leaders who were pushed for an
all-inclusive process putting the pharmaceuticals industry in public context, more and more
securely and quickly. Futuristic regulatory standardization in between the agencies is vital to
minimize redundancy and also accelerate review process in workload with regards to
stakeholders’ benefits.
39
Secure Best Marks with AI Grader
Need help grading? Try our AI Grader for instant feedback on your assignments.

Chapter 5 : Conclusion and recommendations
The FDA and EMA share a series of resemblances and differences as described in the
above research study. Although the centralized, decentralized and mutual recognition
procedure plays a very important role in drug application, market approval, market
distribution, delivery of the drug products, but there exists various gaps within these
administrative bodies – FDA and EMA which are strikingly similar and dissimilar in many
ways. FDA being the more powerful of the two authorities, play a huge role in approval and
distribution of new drugs across united states of America. While on the other hand, European
me4dical agency also known as EMA play a subordinate part to the European Union and
other administrative bodies. So, it can concluded that EMA, as a policymaker and decision
maker is still behind FDA, in terms of the legislative and executive powers – one exercises
over a nation. The study emphases on the fact that FDA being an older administrative body in
the business has been more years and the years of market and research experience has helped
them evolve over the last 40 to 50 years but applying the same experiential theory of
evolution to EMA, the findings are rather murkier. FDA has grown throughout the years
owing to its governmental support and the evolution of national framework with global
business but on the other arm, EMA still remains in lower echelon to the high drug approval
bodies and this leads to long wait time in approval of oncology and other drugs at the very
national level.
Any new drug is hope to a set of patients who cannot be cured by any other drugs and
such is the case in cancer. Cancer often referred to as the malignant disease due its fast
metastasis and malignancy, especially to a level of dread, when it starts to involve or infiltrate
40
The FDA and EMA share a series of resemblances and differences as described in the
above research study. Although the centralized, decentralized and mutual recognition
procedure plays a very important role in drug application, market approval, market
distribution, delivery of the drug products, but there exists various gaps within these
administrative bodies – FDA and EMA which are strikingly similar and dissimilar in many
ways. FDA being the more powerful of the two authorities, play a huge role in approval and
distribution of new drugs across united states of America. While on the other hand, European
me4dical agency also known as EMA play a subordinate part to the European Union and
other administrative bodies. So, it can concluded that EMA, as a policymaker and decision
maker is still behind FDA, in terms of the legislative and executive powers – one exercises
over a nation. The study emphases on the fact that FDA being an older administrative body in
the business has been more years and the years of market and research experience has helped
them evolve over the last 40 to 50 years but applying the same experiential theory of
evolution to EMA, the findings are rather murkier. FDA has grown throughout the years
owing to its governmental support and the evolution of national framework with global
business but on the other arm, EMA still remains in lower echelon to the high drug approval
bodies and this leads to long wait time in approval of oncology and other drugs at the very
national level.
Any new drug is hope to a set of patients who cannot be cured by any other drugs and
such is the case in cancer. Cancer often referred to as the malignant disease due its fast
metastasis and malignancy, especially to a level of dread, when it starts to involve or infiltrate
40

the vital organs of the body and then the connective tissue. A cancer seeding through blood
would spread faster than a cancer spreading through lymph and this speed of metastasis often
determines the level of severity and affliction of an affected patient. When the symptoms are
hard to control, it leads to other problems and multisystem failure in the end. To prevent this,
curing the root of the cause, that is the etiology of the disease is and should always be
preferred over symptomatic management. In cancer, medicine or medical management play a
critical role as compared to surgical management. While drugs are of utmost importance that
decides life and death of the patient – the problems starts. It begins with the myriad of issues
that are associated with the drug discovery and skilled scientists are hard to acquire. Lack of
training and knowledge and lack of market research and feedback causes a gap between the
demands and the ideas conceived to design an appropriate drug to handle a specific type of
cancer. Even with the right workforce and correct designing of the drug – the second problem
that stands up is the approval of the designed or discovered drug which has to be aligned with
the safety and risk measures of the patient situations.
While drug approval involves a crucial and complex series of approvals done both
scientifically and legislatively – it also needs the right heads and right minds to focus on the
right perspectives to focus on the same stages of drug approval. FDA does it very rapidly
while EMA performs the drug approval process for a very prolonged period of time and both
are dangerous as well as cost ineffective. As discussed in the study, that randomized control
trials is extremely important to determine the level of safety and effectiveness a drug provides
and this safety – effectiveness ratio is important to be determined. Secondary researches and
drug approval without RCT has been the major problem with both FDA and EMA which has
been discussed throughout the paper. The critical and most cardinal aspect of leaving out
Randomized control trials or human control trials is extra consumption of time and the fear or
lack of confidence that a drug which is designed with so much effort might not be as effective
41
would spread faster than a cancer spreading through lymph and this speed of metastasis often
determines the level of severity and affliction of an affected patient. When the symptoms are
hard to control, it leads to other problems and multisystem failure in the end. To prevent this,
curing the root of the cause, that is the etiology of the disease is and should always be
preferred over symptomatic management. In cancer, medicine or medical management play a
critical role as compared to surgical management. While drugs are of utmost importance that
decides life and death of the patient – the problems starts. It begins with the myriad of issues
that are associated with the drug discovery and skilled scientists are hard to acquire. Lack of
training and knowledge and lack of market research and feedback causes a gap between the
demands and the ideas conceived to design an appropriate drug to handle a specific type of
cancer. Even with the right workforce and correct designing of the drug – the second problem
that stands up is the approval of the designed or discovered drug which has to be aligned with
the safety and risk measures of the patient situations.
While drug approval involves a crucial and complex series of approvals done both
scientifically and legislatively – it also needs the right heads and right minds to focus on the
right perspectives to focus on the same stages of drug approval. FDA does it very rapidly
while EMA performs the drug approval process for a very prolonged period of time and both
are dangerous as well as cost ineffective. As discussed in the study, that randomized control
trials is extremely important to determine the level of safety and effectiveness a drug provides
and this safety – effectiveness ratio is important to be determined. Secondary researches and
drug approval without RCT has been the major problem with both FDA and EMA which has
been discussed throughout the paper. The critical and most cardinal aspect of leaving out
Randomized control trials or human control trials is extra consumption of time and the fear or
lack of confidence that a drug which is designed with so much effort might not be as effective
41

as one would have thought or assumed previously. This creates a huge perceptional block
whether to be using Randomized control trial or not to be using human clinical trials in the
approval validation process. This precedes the market approval process and is often
concerned with the pre-clinical methodologies that are to be followed imperatively and very
pertinently perhaps. But in cases of both FDA and EMA – it mostly lack the most essential
component of the approval process that is randomized control testing which is important for
understanding the real world and real application of a proposed drug. While most of the drugs
approved in the United States market, do not have RCT test result for it was never conducted
and as a matter of fact – the drugs without RCT results are continuing their course in the
market for more than 5 years. The feedback, from the hospitals and the medical setting where
the drug has been used, although not very satisfactory or not at all satisfactory – are
continued in the markets by FDA and no measures for withdrawal are taken. This study
concludes this unchartered market availability of non RCT validated drugs is a major barrier
in terms of both threat to human lives and social perception of oncology drugs – the oncology
drug poses.
EMA on the other hand, after having approved a drug cannot still distribute them to
the markets in United Kingdom and it leads to a lot of lead in contrast to the ‘accelerated drug
approval process of the FDA. The member states are too large in numbers than the authority
processes in United kingdom and the broad spectrum of laws, rules and regulation that cross
functions in approval of a single oncology make the process of oncology drug approval,
painfully longer. While the EMA tends to learn from the feedbacks of FDA approved drugs
and the FDA tends to learn from the system of EMA on a vice versa note. But the study
points out that the connections between these two powerful agencies of global drug market –
is still fragile and there exists a huge scope for growth and development in terms of cross
nation communication and collective research approaches and societal and medical
42
whether to be using Randomized control trial or not to be using human clinical trials in the
approval validation process. This precedes the market approval process and is often
concerned with the pre-clinical methodologies that are to be followed imperatively and very
pertinently perhaps. But in cases of both FDA and EMA – it mostly lack the most essential
component of the approval process that is randomized control testing which is important for
understanding the real world and real application of a proposed drug. While most of the drugs
approved in the United States market, do not have RCT test result for it was never conducted
and as a matter of fact – the drugs without RCT results are continuing their course in the
market for more than 5 years. The feedback, from the hospitals and the medical setting where
the drug has been used, although not very satisfactory or not at all satisfactory – are
continued in the markets by FDA and no measures for withdrawal are taken. This study
concludes this unchartered market availability of non RCT validated drugs is a major barrier
in terms of both threat to human lives and social perception of oncology drugs – the oncology
drug poses.
EMA on the other hand, after having approved a drug cannot still distribute them to
the markets in United Kingdom and it leads to a lot of lead in contrast to the ‘accelerated drug
approval process of the FDA. The member states are too large in numbers than the authority
processes in United kingdom and the broad spectrum of laws, rules and regulation that cross
functions in approval of a single oncology make the process of oncology drug approval,
painfully longer. While the EMA tends to learn from the feedbacks of FDA approved drugs
and the FDA tends to learn from the system of EMA on a vice versa note. But the study
points out that the connections between these two powerful agencies of global drug market –
is still fragile and there exists a huge scope for growth and development in terms of cross
nation communication and collective research approaches and societal and medical
42
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

institutional problem solving. As secondary researches saves time and aid in faster
manufacturing of an oncology drug but this this can still be dreadful under the circumstances
that a drug approved by the scientists and systems of a nation is based on the characteristics
of population studies performed within the nation and it might have a different impact on a
different population and this is where, according to this research study, the implication lies.
The limitation of this study lies in the fact that we studied the procedural differences and
similarities between FDA and EMA very deeply while some of the internal administrative
function could not be covered due to lack of such information across the available scholarly
resources. The implication of this study stresses on the fact that more researches should be
taken up in order to understand the internal socioeconomic dynamics that works truly on a
pragmatic axis behind an embellished business or trade front. This information would
complement the aforementioned findings of this research. The revelations of this research
study can be transferred to higher forums to confirm and validate our results further so that it
can be incorporated in curriculum of educational settings.
The study recommends that the communication gaps on a global forum and
specifically, the gaps between the researches undertaken by FDA and EMA has to be bridged
pertinently. While, the FDA needs to slow down a bit, giving apt amount of time to each step
and stage of drug manufacturing process, the EMA on the other hand must speed up the drug
approval process as not to keep the market waiting. But in either of the cases – the oncology
drugs must go through a proper human clinical trial and randomized control trial validation.
Proper documentation of each of the drug approval process should be made mandatory by
both FDA and EMA and feedbacks regarding the efficiency of the drugs must be dealt with
seriously by both these mighty agencies. The study suggests that in order to reduce the
expense of individual human clinical trials and randomized control trials – the two agencies
FDA and EMA can take up joint approaches nevertheless to validate their drugs with a proper
43
manufacturing of an oncology drug but this this can still be dreadful under the circumstances
that a drug approved by the scientists and systems of a nation is based on the characteristics
of population studies performed within the nation and it might have a different impact on a
different population and this is where, according to this research study, the implication lies.
The limitation of this study lies in the fact that we studied the procedural differences and
similarities between FDA and EMA very deeply while some of the internal administrative
function could not be covered due to lack of such information across the available scholarly
resources. The implication of this study stresses on the fact that more researches should be
taken up in order to understand the internal socioeconomic dynamics that works truly on a
pragmatic axis behind an embellished business or trade front. This information would
complement the aforementioned findings of this research. The revelations of this research
study can be transferred to higher forums to confirm and validate our results further so that it
can be incorporated in curriculum of educational settings.
The study recommends that the communication gaps on a global forum and
specifically, the gaps between the researches undertaken by FDA and EMA has to be bridged
pertinently. While, the FDA needs to slow down a bit, giving apt amount of time to each step
and stage of drug manufacturing process, the EMA on the other hand must speed up the drug
approval process as not to keep the market waiting. But in either of the cases – the oncology
drugs must go through a proper human clinical trial and randomized control trial validation.
Proper documentation of each of the drug approval process should be made mandatory by
both FDA and EMA and feedbacks regarding the efficiency of the drugs must be dealt with
seriously by both these mighty agencies. The study suggests that in order to reduce the
expense of individual human clinical trials and randomized control trials – the two agencies
FDA and EMA can take up joint approaches nevertheless to validate their drugs with a proper
43

Randomized control trial. Workforce training and education is critical to designing of an
suitable drug that fits the expectations and aim of the drug discovery proper or the vision held
at the very beginning.
REFERENCES.
1. Ades, F., Senterre, C., Zardavas, D., de Azambuja, E., Popescu, R. and Piccart, M.,
2017. Are life-saving anticancer drugs reaching all patients? Patterns and
discrepancies of trastuzumab use in the European Union and the USA. PloS one,
12(3), p.e0172351.
2. Arnould, B., Acquadro, C., Lanar, S. and Savre, I., 2018. Role of Patient-Reported
Outcome Evaluation in the Approval of Orphan Drugs: A Review of 15 Year
Approvals by the FDA and the EMA. Value in Health, 21, p.S257.
3. Austin, C.P., Cutillo, C.M., Lau, L.P., Jonker, A.H., Rath, A., Julkowska, D.,
Thomson, D., Terry, S.F., de Montleau, B., Ardigò, D. and Hivert, V., 2018. Future of
rare diseases research 2017–2027: an IRDiRC perspective. Clinical and translational
science, 11(1), pp.21-27.
4. Brunault, P., Champagne, A.L., Huguet, G., Suzanne, I., Senon, J.L., Body, G.,
Rusch, E., Magnin, G., Voyer, M., Réveillère, C. and Camus, V., 2016. Major
depressive disorder, personality disorders, and coping strategies are independent risk
factors for lower quality of life in non‐metastatic breast cancer patients. Psycho
‐
Oncology, 25(5), pp.513-520.
5. Cardot, J.M., Arieta, A.G., Paixao, P., Tasevska, I. and Davit, B., 2016. Implementing
the biopharmaceutics classification system in drug development: reconciling
44
suitable drug that fits the expectations and aim of the drug discovery proper or the vision held
at the very beginning.
REFERENCES.
1. Ades, F., Senterre, C., Zardavas, D., de Azambuja, E., Popescu, R. and Piccart, M.,
2017. Are life-saving anticancer drugs reaching all patients? Patterns and
discrepancies of trastuzumab use in the European Union and the USA. PloS one,
12(3), p.e0172351.
2. Arnould, B., Acquadro, C., Lanar, S. and Savre, I., 2018. Role of Patient-Reported
Outcome Evaluation in the Approval of Orphan Drugs: A Review of 15 Year
Approvals by the FDA and the EMA. Value in Health, 21, p.S257.
3. Austin, C.P., Cutillo, C.M., Lau, L.P., Jonker, A.H., Rath, A., Julkowska, D.,
Thomson, D., Terry, S.F., de Montleau, B., Ardigò, D. and Hivert, V., 2018. Future of
rare diseases research 2017–2027: an IRDiRC perspective. Clinical and translational
science, 11(1), pp.21-27.
4. Brunault, P., Champagne, A.L., Huguet, G., Suzanne, I., Senon, J.L., Body, G.,
Rusch, E., Magnin, G., Voyer, M., Réveillère, C. and Camus, V., 2016. Major
depressive disorder, personality disorders, and coping strategies are independent risk
factors for lower quality of life in non‐metastatic breast cancer patients. Psycho
‐
Oncology, 25(5), pp.513-520.
5. Cardot, J.M., Arieta, A.G., Paixao, P., Tasevska, I. and Davit, B., 2016. Implementing
the biopharmaceutics classification system in drug development: reconciling
44

similarities, differences, and shared challenges in the EMA and US-FDA-
recommended approaches. The AAPS journal, 18(4), pp.1039-1046.
6. Corsello, S.M., Bittker, J.A., Liu, Z., Gould, J., McCarren, P., Hirschman, J.E.,
Johnston, S.E., Vrcic, A., Wong, B., Khan, M. and Asiedu, J., 2017. The Drug
Repurposing Hub: a next-generation drug library and information resource. Nature
medicine, 23(4), p.405.
7. Davis, C., Naci, H., Gurpinar, E., Poplavska, E., Pinto, A. and Aggarwal, A., 2017.
Availability of evidence of benefits on overall survival and quality of life of cancer
drugs approved by European Medicines Agency: retrospective cohort study of drug
approvals 2009-13. bmj, 359, p.j4530.
8. Davis, C., Naci, H., Gurpinar, E., Poplavska, E., Pinto, A. and Aggarwal, A., 2017.
Availability of evidence of benefits on overall survival and quality of life of cancer
drugs approved by European Medicines Agency: retrospective cohort study of drug
approvals 2009-13. bmj, 359, p.j4530.
9. Davit, B.M., Kanfer, I., Tsang, Y.C. and Cardot, J.M., 2016. BCS biowaivers:
similarities and differences among EMA, FDA, and WHO requirements. The AAPS
journal, 18(3), pp.612-618.
10. Desfontaine, V., Guillarme, D., Francotte, E. and Nováková, L., 2015. Supercritical
fluid chromatography in pharmaceutical analysis. Journal of pharmaceutical and
biomedical analysis, 113, pp.56-71.
11. Dowell, D., Haegerich, T.M. and Chou, R., 2016. CDC guideline for prescribing
opioids for chronic pain—United States, 2016. Jama, 315(15), pp.1624-1645.
12. Eder, C. and Wild, C., 2019. Technology forecast: advanced therapies in late clinical
research, EMA approval or clinical application via hospital exemption. Journal of
market access & health policy, 7(1), p.1600939.
45
recommended approaches. The AAPS journal, 18(4), pp.1039-1046.
6. Corsello, S.M., Bittker, J.A., Liu, Z., Gould, J., McCarren, P., Hirschman, J.E.,
Johnston, S.E., Vrcic, A., Wong, B., Khan, M. and Asiedu, J., 2017. The Drug
Repurposing Hub: a next-generation drug library and information resource. Nature
medicine, 23(4), p.405.
7. Davis, C., Naci, H., Gurpinar, E., Poplavska, E., Pinto, A. and Aggarwal, A., 2017.
Availability of evidence of benefits on overall survival and quality of life of cancer
drugs approved by European Medicines Agency: retrospective cohort study of drug
approvals 2009-13. bmj, 359, p.j4530.
8. Davis, C., Naci, H., Gurpinar, E., Poplavska, E., Pinto, A. and Aggarwal, A., 2017.
Availability of evidence of benefits on overall survival and quality of life of cancer
drugs approved by European Medicines Agency: retrospective cohort study of drug
approvals 2009-13. bmj, 359, p.j4530.
9. Davit, B.M., Kanfer, I., Tsang, Y.C. and Cardot, J.M., 2016. BCS biowaivers:
similarities and differences among EMA, FDA, and WHO requirements. The AAPS
journal, 18(3), pp.612-618.
10. Desfontaine, V., Guillarme, D., Francotte, E. and Nováková, L., 2015. Supercritical
fluid chromatography in pharmaceutical analysis. Journal of pharmaceutical and
biomedical analysis, 113, pp.56-71.
11. Dowell, D., Haegerich, T.M. and Chou, R., 2016. CDC guideline for prescribing
opioids for chronic pain—United States, 2016. Jama, 315(15), pp.1624-1645.
12. Eder, C. and Wild, C., 2019. Technology forecast: advanced therapies in late clinical
research, EMA approval or clinical application via hospital exemption. Journal of
market access & health policy, 7(1), p.1600939.
45
Secure Best Marks with AI Grader
Need help grading? Try our AI Grader for instant feedback on your assignments.

13. Fontes Jardim, D.L., Schwaederle, M., Wei, C., Lee, J.J., Hong, D.S., Eggermont,
A.M., Schilsky, R.L., Mendelsohn, J., Lazar, V. and Kurzrock, R., 2015. Impact of a
biomarker-based strategy on oncology drug development: a meta-analysis of clinical
trials leading to FDA approval. JNCI: Journal of the National Cancer
Institute, 107(11).
14. Forli, S., Huey, R., Pique, M.E., Sanner, M.F., Goodsell, D.S. and Olson, A.J., 2016.
Computational protein–ligand docking and virtual drug screening with the AutoDock
suite. Nature protocols, 11(5), p.905.
15. Frye, S.V., Arkin, M.R., Arrowsmith, C.H., Conn, P.J., Glicksman, M.A., Hull-Ryde,
E.A. and Slusher, B.S., 2015. Tackling reproducibility in academic preclinical drug
discovery. Nature Reviews Drug Discovery, 14(11), p.733.
16. Gammie, T., Lu, C.Y. and Babar, Z.U.D., 2015. Access to orphan drugs: a
comprehensive review of legislations, regulations and policies in 35 countries. PloS
one, 10(10), p.e0140002.
17. Hochhaus, A., Larson, R.A., Guilhot, F., Radich, J.P., Branford, S., Hughes, T.P.,
Baccarani, M., Deininger, M.W., Cervantes, F., Fujihara, S. and Ortmann, C.E., 2017.
Long-term outcomes of imatinib treatment for chronic myeloid leukemia. New
England Journal of Medicine, 376(10), pp.917-927.
18. Jasińska-Stroschein, M., Kurczewska, U. and Orszulak-Michalak, D., 2017. Statistical
Considerations Concerning Dissimilar Regulatory Requirements for Dissolution
Similarity Assessment. The Example of Immediate-Release Dosage Forms. Journal of
pharmaceutical sciences, 106(5), pp.1275-1284.
19. Jonsson, B., Martinalbo, J. and Pignatti, F., 2017. European Medicines Agency
Perspective on Oncology Study Design for Marketing Authorization and Beyond.
Clinical Pharmacology & Therapeutics, 101(5), pp.577-579.
46
A.M., Schilsky, R.L., Mendelsohn, J., Lazar, V. and Kurzrock, R., 2015. Impact of a
biomarker-based strategy on oncology drug development: a meta-analysis of clinical
trials leading to FDA approval. JNCI: Journal of the National Cancer
Institute, 107(11).
14. Forli, S., Huey, R., Pique, M.E., Sanner, M.F., Goodsell, D.S. and Olson, A.J., 2016.
Computational protein–ligand docking and virtual drug screening with the AutoDock
suite. Nature protocols, 11(5), p.905.
15. Frye, S.V., Arkin, M.R., Arrowsmith, C.H., Conn, P.J., Glicksman, M.A., Hull-Ryde,
E.A. and Slusher, B.S., 2015. Tackling reproducibility in academic preclinical drug
discovery. Nature Reviews Drug Discovery, 14(11), p.733.
16. Gammie, T., Lu, C.Y. and Babar, Z.U.D., 2015. Access to orphan drugs: a
comprehensive review of legislations, regulations and policies in 35 countries. PloS
one, 10(10), p.e0140002.
17. Hochhaus, A., Larson, R.A., Guilhot, F., Radich, J.P., Branford, S., Hughes, T.P.,
Baccarani, M., Deininger, M.W., Cervantes, F., Fujihara, S. and Ortmann, C.E., 2017.
Long-term outcomes of imatinib treatment for chronic myeloid leukemia. New
England Journal of Medicine, 376(10), pp.917-927.
18. Jasińska-Stroschein, M., Kurczewska, U. and Orszulak-Michalak, D., 2017. Statistical
Considerations Concerning Dissimilar Regulatory Requirements for Dissolution
Similarity Assessment. The Example of Immediate-Release Dosage Forms. Journal of
pharmaceutical sciences, 106(5), pp.1275-1284.
19. Jonsson, B., Martinalbo, J. and Pignatti, F., 2017. European Medicines Agency
Perspective on Oncology Study Design for Marketing Authorization and Beyond.
Clinical Pharmacology & Therapeutics, 101(5), pp.577-579.
46

20. Katsuno, K., Burrows, J.N., Duncan, K., Van Huijsduijnen, R.H., Kaneko, T., Kita,
K., Mowbray, C.E., Schmatz, D., Warner, P. and Slingsby, B.T., 2015. Hit and lead
criteria in drug discovery for infectious diseases of the developing world. Nature
Reviews drug discovery, 14(11), p.751.
21. Koinig, I., Diehl, S. and Mueller, B., 2018. Exploring antecedents of attitudes and
skepticism towards pharmaceutical advertising and inter-attitudinal and inter-
skepticism consistency on three levels: an international study. International Journal
of Advertising, 37(5), pp.718-748.
22. Kunz, M. and Hölzel, M., 2017. The impact of melanoma genetics on treatment
response and resistance in clinical and experimental studies. Cancer and Metastasis
Reviews, 36(1), pp.537
23. Lagassé, H.D., Alexaki, A., Simhadri, V.L., Katagiri, N.H., Jankowski, W., Sauna,
Z.E. and Kimchi-Sarfaty, C., 2017. Recent advances in (therapeutic protein) drug
development. F1000Research, 6.
24. LeBeau, A.P., 2019. Considerations for implementing an informatics system to
support biologics drug discovery. Drug discovery today, 24(1), pp.42-45.
25. Melmed, S., 2017. Acromegaly. In The pituitary (pp. 423-466). Academic Press.
26. Ozsvári, B., Lamb, R. and Lisanti, M.P., 2016. Repurposing of FDA-approved drugs
against cancer–Focus on metastasis. Aging (Albany NY), 8(4), p.567.
27. Paglialunga, S., Offman, E., Ichhpurani, N., Marbury, T.C. and Morimoto, B.H.,
2017. Update and trends on pharmacokinetic studies in patients with impaired renal
function: practical insight into application of the FDA and EMA guidelines. Expert
review of clinical pharmacology, 10(3), pp.273-283.
47
K., Mowbray, C.E., Schmatz, D., Warner, P. and Slingsby, B.T., 2015. Hit and lead
criteria in drug discovery for infectious diseases of the developing world. Nature
Reviews drug discovery, 14(11), p.751.
21. Koinig, I., Diehl, S. and Mueller, B., 2018. Exploring antecedents of attitudes and
skepticism towards pharmaceutical advertising and inter-attitudinal and inter-
skepticism consistency on three levels: an international study. International Journal
of Advertising, 37(5), pp.718-748.
22. Kunz, M. and Hölzel, M., 2017. The impact of melanoma genetics on treatment
response and resistance in clinical and experimental studies. Cancer and Metastasis
Reviews, 36(1), pp.537
23. Lagassé, H.D., Alexaki, A., Simhadri, V.L., Katagiri, N.H., Jankowski, W., Sauna,
Z.E. and Kimchi-Sarfaty, C., 2017. Recent advances in (therapeutic protein) drug
development. F1000Research, 6.
24. LeBeau, A.P., 2019. Considerations for implementing an informatics system to
support biologics drug discovery. Drug discovery today, 24(1), pp.42-45.
25. Melmed, S., 2017. Acromegaly. In The pituitary (pp. 423-466). Academic Press.
26. Ozsvári, B., Lamb, R. and Lisanti, M.P., 2016. Repurposing of FDA-approved drugs
against cancer–Focus on metastasis. Aging (Albany NY), 8(4), p.567.
27. Paglialunga, S., Offman, E., Ichhpurani, N., Marbury, T.C. and Morimoto, B.H.,
2017. Update and trends on pharmacokinetic studies in patients with impaired renal
function: practical insight into application of the FDA and EMA guidelines. Expert
review of clinical pharmacology, 10(3), pp.273-283.
47

28. Pascoalino, B.S., Courtemanche, G., Cordeiro, M.T., Gil, L.H. and Freitas-Junior, L.,
2016. Zika antiviral chemotherapy: identification of drugs and promising starting
points for drug discovery from an FDA-approved library. F1000Research, 5.
29. Prigerson, H.G., Bao, Y., Shah, M.A., Paulk, M.E., LeBlanc, T.W., Schneider, B.J.,
Garrido, M.M., Reid, M.C., Berlin, D.A., Adelson, K.B. and Neugut, A.I., 2015.
Chemotherapy use, performance status, and quality of life at the end of life. JAMA
oncology, 1(6), pp.778-784.
30. Rodieux, F., Storelli, F., Manzano, S., Gervaix, A., Posfay-Barbe, K., Samer, C.,
Daali, Y. and Desmeules, J., 2019. P83 CYP2D6 phenotyping in children treated with
tramadol at the Emergency Department.
31. Rose, K., 2019. P84 Pediatric oncology studies triggered by the united states (US)
food and drug administration (FDA) and the european union (EU) european
medicines agency (EMA) aim at labels, not at improved treatment. Some harm young
patients by exposing them to substandard monotherapy instead of combination
treatment.
32. Saber, H. and Leighton, J.K., 2015. An FDA oncology analysis of antibody-drug
conjugates. Regulatory Toxicology and Pharmacology, 71(3), pp.444-452.
33. Schnipper, L.E., Davidson, N.E., Wollins, D.S., Blayney, D.W., Dicker, A.P., Ganz,
P.A., Hoverman, J.R., Langdon, R., Lyman, G.H., Meropol, N.J. and Mulvey, T.,
2016. Updating the American Society of Clinical Oncology value framework:
revisions and reflections in response to comments received. Journal of Clinical
Oncology, 34(24), pp.2925-2934.
34. Shields, R.K., Potoski, B.A., Haidar, G., Hao, B., Doi, Y., Chen, L., Press, E.G.,
Kreiswirth, B.N., Clancy, C.J. and Nguyen, M.H., 2016. Clinical outcomes, drug
toxicity, and emergence of ceftazidime-avibactam resistance among patients treated
48
2016. Zika antiviral chemotherapy: identification of drugs and promising starting
points for drug discovery from an FDA-approved library. F1000Research, 5.
29. Prigerson, H.G., Bao, Y., Shah, M.A., Paulk, M.E., LeBlanc, T.W., Schneider, B.J.,
Garrido, M.M., Reid, M.C., Berlin, D.A., Adelson, K.B. and Neugut, A.I., 2015.
Chemotherapy use, performance status, and quality of life at the end of life. JAMA
oncology, 1(6), pp.778-784.
30. Rodieux, F., Storelli, F., Manzano, S., Gervaix, A., Posfay-Barbe, K., Samer, C.,
Daali, Y. and Desmeules, J., 2019. P83 CYP2D6 phenotyping in children treated with
tramadol at the Emergency Department.
31. Rose, K., 2019. P84 Pediatric oncology studies triggered by the united states (US)
food and drug administration (FDA) and the european union (EU) european
medicines agency (EMA) aim at labels, not at improved treatment. Some harm young
patients by exposing them to substandard monotherapy instead of combination
treatment.
32. Saber, H. and Leighton, J.K., 2015. An FDA oncology analysis of antibody-drug
conjugates. Regulatory Toxicology and Pharmacology, 71(3), pp.444-452.
33. Schnipper, L.E., Davidson, N.E., Wollins, D.S., Blayney, D.W., Dicker, A.P., Ganz,
P.A., Hoverman, J.R., Langdon, R., Lyman, G.H., Meropol, N.J. and Mulvey, T.,
2016. Updating the American Society of Clinical Oncology value framework:
revisions and reflections in response to comments received. Journal of Clinical
Oncology, 34(24), pp.2925-2934.
34. Shields, R.K., Potoski, B.A., Haidar, G., Hao, B., Doi, Y., Chen, L., Press, E.G.,
Kreiswirth, B.N., Clancy, C.J. and Nguyen, M.H., 2016. Clinical outcomes, drug
toxicity, and emergence of ceftazidime-avibactam resistance among patients treated
48
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

for carbapenem-resistant Enterobacteriaceae infections. Clinical Infectious
Diseases, 63(12), pp.1615-1618.
35. Sifuentes, M.M. and Giuffrida, A., 2015. Drug review differences across the United
States and the European Union. Pharmaceutical Regulatory Affairs: Open Access, 4,
p.e156.
36. Strouse, J.J., Fears, T.R., Tucker, M.A. and Wayne, A.S., 2005. Pediatric melanoma:
risk factor and survival analysis of the surveillance, epidemiology and end results
database. Journal of Clinical Oncology, 23(21), pp.4735-4741.
37. Surmeier, D.J., Day, M., Ding, J., Chan, C.S., Guzman, J., Mercer, J., Tkatch, T.,
Wang, Z. and Ilijic, E., Northwestern University, 2018. Methods and compositions for
modulating calcium channels. U.S. Patent Application 16/002,626.
38. Vivot, A., Jacot, J., Zeitoun, J.D., Ravaud, P., Crequit, P. and Porcher, R., 2017.
Clinical benefit, price and approval characteristics of FDA-approved new drugs for
treating advanced solid cancer, 2000–2015. Annals of Oncology, 28(5), pp.1111-1116.
39. Wainwright, C.E., Elborn, J.S., Ramsey, B.W., Marigowda, G., Huang, X., Cipolli,
M., Colombo, C., Davies, J.C., De Boeck, K., Flume, P.A. and Konstan, M.W., 2015.
Lumacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del
CFTR. New England Journal of Medicine, 373(3), pp.220-231. 5.
40. Wallwiener, M., Matthies, L., Simoes, E., Keilmann, L., Hartkopf, A.D., Sokolov,
A.N., Walter, C.B., Sickenberger, N., Wallwiener, S., Feisst, M. and Gass, P., 2017.
Reliability of an e-PRO tool of EORTC QLQ-C30 for measurement of health-related
quality of life in patients with breast cancer: prospective randomized trial. Journal of
medical Internet research, 19(9), p.e322.
41. Wdlff, J.A., 1991. History of pediatric oncology. Pediatric hematology and
oncology, 8(2), pp.89-91.
49
Diseases, 63(12), pp.1615-1618.
35. Sifuentes, M.M. and Giuffrida, A., 2015. Drug review differences across the United
States and the European Union. Pharmaceutical Regulatory Affairs: Open Access, 4,
p.e156.
36. Strouse, J.J., Fears, T.R., Tucker, M.A. and Wayne, A.S., 2005. Pediatric melanoma:
risk factor and survival analysis of the surveillance, epidemiology and end results
database. Journal of Clinical Oncology, 23(21), pp.4735-4741.
37. Surmeier, D.J., Day, M., Ding, J., Chan, C.S., Guzman, J., Mercer, J., Tkatch, T.,
Wang, Z. and Ilijic, E., Northwestern University, 2018. Methods and compositions for
modulating calcium channels. U.S. Patent Application 16/002,626.
38. Vivot, A., Jacot, J., Zeitoun, J.D., Ravaud, P., Crequit, P. and Porcher, R., 2017.
Clinical benefit, price and approval characteristics of FDA-approved new drugs for
treating advanced solid cancer, 2000–2015. Annals of Oncology, 28(5), pp.1111-1116.
39. Wainwright, C.E., Elborn, J.S., Ramsey, B.W., Marigowda, G., Huang, X., Cipolli,
M., Colombo, C., Davies, J.C., De Boeck, K., Flume, P.A. and Konstan, M.W., 2015.
Lumacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del
CFTR. New England Journal of Medicine, 373(3), pp.220-231. 5.
40. Wallwiener, M., Matthies, L., Simoes, E., Keilmann, L., Hartkopf, A.D., Sokolov,
A.N., Walter, C.B., Sickenberger, N., Wallwiener, S., Feisst, M. and Gass, P., 2017.
Reliability of an e-PRO tool of EORTC QLQ-C30 for measurement of health-related
quality of life in patients with breast cancer: prospective randomized trial. Journal of
medical Internet research, 19(9), p.e322.
41. Wdlff, J.A., 1991. History of pediatric oncology. Pediatric hematology and
oncology, 8(2), pp.89-91.
49

42. Zeitoun, J.D., Baron, G., Vivot, A., Atal, I., Downing, N.S., Ross, J.S. and Ravaud,
P., 2018. Post‐marketing research and its outcome for novel anticancer agents
approved by both the FDA and EMA between 2005 and 2010: A cross‐sectional
study. International journal of cancer, 142(2), pp.414-423.
43. Morgenthau, H. J. 2017. Positivism, functionalism, and international law. In The
Nature of International Law (pp. 159-184). Routledge.
44. Pierre, E.A.S., 2016. The long reach of logical positivism/logical empiricism.
In Qualitative inquiry through a critical lens (pp. 27-38). Routledge.
45. Sullivan, H., 2016. Interpretivism and Public Policy Research. In Interpreting
Governance, High Politics, and Public Policy(pp. 184-204). Routledge.
46. Hartwig, M., 2015. Dictionary of critical realism. Routledge.
47. Rysiew, P., 2017. A case for direct realism?. In New Essays on Thomas Reid (pp. 51-
56). Routledge.
48. McAbee, S.T., Landis, R.S. and Burke, M.I., 2017. Inductive reasoning: The promise
of big data. Human Resource Management Review, 27(2), pp.277-290.
49. Alfred, K.L., Connolly, A.C. and Kraemer, D.J., 2018. Putting the pieces together:
generating a novel representational space through deductive reasoning. NeuroImage,
183, pp.99-111.
50. Dunne, D.D. and Dougherty, D., 2016. Abductive reasoning: How innovators
navigate in the labyrinth of complex product innovation. Organization Studies, 37(2),
pp.131-159.
51. Claessen, M.H., van der Ham, I.J., Jagersma, E. and Visser-Meily, J.M., 2016.
Navigation strategy training using virtual reality in six chronic stroke patients: a novel
and explorative approach to the rehabilitation of navigation impairment.
Neuropsychological rehabilitation, 26(5-6), pp.822-846.
50
P., 2018. Post‐marketing research and its outcome for novel anticancer agents
approved by both the FDA and EMA between 2005 and 2010: A cross‐sectional
study. International journal of cancer, 142(2), pp.414-423.
43. Morgenthau, H. J. 2017. Positivism, functionalism, and international law. In The
Nature of International Law (pp. 159-184). Routledge.
44. Pierre, E.A.S., 2016. The long reach of logical positivism/logical empiricism.
In Qualitative inquiry through a critical lens (pp. 27-38). Routledge.
45. Sullivan, H., 2016. Interpretivism and Public Policy Research. In Interpreting
Governance, High Politics, and Public Policy(pp. 184-204). Routledge.
46. Hartwig, M., 2015. Dictionary of critical realism. Routledge.
47. Rysiew, P., 2017. A case for direct realism?. In New Essays on Thomas Reid (pp. 51-
56). Routledge.
48. McAbee, S.T., Landis, R.S. and Burke, M.I., 2017. Inductive reasoning: The promise
of big data. Human Resource Management Review, 27(2), pp.277-290.
49. Alfred, K.L., Connolly, A.C. and Kraemer, D.J., 2018. Putting the pieces together:
generating a novel representational space through deductive reasoning. NeuroImage,
183, pp.99-111.
50. Dunne, D.D. and Dougherty, D., 2016. Abductive reasoning: How innovators
navigate in the labyrinth of complex product innovation. Organization Studies, 37(2),
pp.131-159.
51. Claessen, M.H., van der Ham, I.J., Jagersma, E. and Visser-Meily, J.M., 2016.
Navigation strategy training using virtual reality in six chronic stroke patients: a novel
and explorative approach to the rehabilitation of navigation impairment.
Neuropsychological rehabilitation, 26(5-6), pp.822-846.
50

52. Abderma, A., Hilal, R. and Beneserighe, D., 2018. Project management in Moroccan
higher education: An explanatory approach according to the Grant and Pennypacker
scale [Le management de projets dans l'enseignement supérieur marocain: Une
approche explicative selon l'échelle de Grant et Pennypacker] (No. hal-01744094).
53. Colorafi, K.J. and Evans, B., 2016. Qualitative descriptive methods in health science
research. HERD: Health Environments Research & Design Journal, 9(4), pp.16-25.
54. Bailey, A.M., Mendicino, M. and Au, P., 2014. An FDA perspective on preclinical
development of cell-based regenerative medicine products. Nature biotechnology,
32(8), p.721.
55. Bobo, D., Robinson, K.J., Islam, J., Thurecht, K.J. and Corrie, S.R., 2016.
Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials
to date. Pharmaceutical research, 33(10), pp.2373-2387.
56. Ciociola, A.A., Cohen, L.B., Kulkarni, P., Kefalas, C., Buchman, A., Burke, C., Cain,
T., Connor, J., Ehrenpreis, E.D., Fang, J. and Fass, R., 2014. How drugs are
developed and approved by the FDA: current process and future directions. The
American journal of gastroenterology, 109(5), p.620.
57. Mullard, A., 2015. 2014 FDA drug approvals.
58. Lawrence, X.Y. and Li, B.V. eds., 2014. FDA bioequivalence standards (Vol. 13).
Springer.
59. Alpern, J.D., Stauffer, W.M. and Kesselheim, A.S., 2014. High-cost generic drugs—
implications for patients and policymakers. New England Journal of Medicine,
371(20), pp.18Elsäßer, A., Regnstrom, J., Vetter, T., Koenig, F., Hemmings, R.J.,
Greco, M., Papaluca-Amati, M. and Posch, M., 2014. Adaptive clinical trial designs
for European marketing authorization: a survey of scientific advice letters from the
European Medicines Agency. Trials, 15(1), p.383.59-1862.
51
higher education: An explanatory approach according to the Grant and Pennypacker
scale [Le management de projets dans l'enseignement supérieur marocain: Une
approche explicative selon l'échelle de Grant et Pennypacker] (No. hal-01744094).
53. Colorafi, K.J. and Evans, B., 2016. Qualitative descriptive methods in health science
research. HERD: Health Environments Research & Design Journal, 9(4), pp.16-25.
54. Bailey, A.M., Mendicino, M. and Au, P., 2014. An FDA perspective on preclinical
development of cell-based regenerative medicine products. Nature biotechnology,
32(8), p.721.
55. Bobo, D., Robinson, K.J., Islam, J., Thurecht, K.J. and Corrie, S.R., 2016.
Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials
to date. Pharmaceutical research, 33(10), pp.2373-2387.
56. Ciociola, A.A., Cohen, L.B., Kulkarni, P., Kefalas, C., Buchman, A., Burke, C., Cain,
T., Connor, J., Ehrenpreis, E.D., Fang, J. and Fass, R., 2014. How drugs are
developed and approved by the FDA: current process and future directions. The
American journal of gastroenterology, 109(5), p.620.
57. Mullard, A., 2015. 2014 FDA drug approvals.
58. Lawrence, X.Y. and Li, B.V. eds., 2014. FDA bioequivalence standards (Vol. 13).
Springer.
59. Alpern, J.D., Stauffer, W.M. and Kesselheim, A.S., 2014. High-cost generic drugs—
implications for patients and policymakers. New England Journal of Medicine,
371(20), pp.18Elsäßer, A., Regnstrom, J., Vetter, T., Koenig, F., Hemmings, R.J.,
Greco, M., Papaluca-Amati, M. and Posch, M., 2014. Adaptive clinical trial designs
for European marketing authorization: a survey of scientific advice letters from the
European Medicines Agency. Trials, 15(1), p.383.59-1862.
51
Secure Best Marks with AI Grader
Need help grading? Try our AI Grader for instant feedback on your assignments.

60. Bonini, S., Eichler, H.G., Wathion, N. and Rasi, G., 2014. Transparency and the
European Medicines Agency—sharing of clinical trial data. New England Journal of
Medicine, 371(26), pp.2452-2455.
61. Alqahtani, S., Seoane‐Vazquez, E., Rodriguez‐Monguio, R. and Eguale, T., 2015.
Priority review drugs approved by the FDA and the EMA: time for international
regulatory harmonization of pharmaceuticals?. Pharmacoepidemiology and drug
safety, 24(7), pp.709-715.
62. Pacurariu, A.C., Coloma, P.M., van Haren, A., Genov, G., Sturkenboom, M.C. and
Straus, S.M., 2014. A description of signals during the first 18 months of the EMA
pharmacovigilance risk assessment committee. Drug safety, 37(12), pp.1059-1066.
63. Vishal, P., Rahulgiri, G., Pratik, M. and Badjatya, J.K., 2014. A review on drug
approval process for US, Europe and India. International Journal of Drug Regulatory
Affairs, 2(1), pp.1-11.
64. Leyens, L. and Brand, A., 2016. Early patient access to medicines: health technology
assessment bodies need to catch up with new marketing authorization methods. Public
Health Genomics, 19(3), pp.187-191.
65. Garattini, L. and Padula, A., 2017. Why EMA should provide clearer guidance on the
authorization of NBCDs in generic and hybrid applications.
66. Monami, M., Dicembrini, I. and Mannucci, E., 2014. Dipeptidyl peptidase‐4
inhibitors and pancreatitis risk: a meta‐analysis of randomized clinical trials. Diabetes,
Obesity and Metabolism, 16(1), pp.48-56.
67. Krcevski‐Skvarc, N., Wells, C. and Häuser, W., 2018. Availability and approval of
cannabis‐based medicines for chronic pain management and palliative/supportive care
in Europe: A survey of the status in the chapters of the European Pain Federation.
European Journal of Pain, 22(3), pp.440-454.
52
European Medicines Agency—sharing of clinical trial data. New England Journal of
Medicine, 371(26), pp.2452-2455.
61. Alqahtani, S., Seoane‐Vazquez, E., Rodriguez‐Monguio, R. and Eguale, T., 2015.
Priority review drugs approved by the FDA and the EMA: time for international
regulatory harmonization of pharmaceuticals?. Pharmacoepidemiology and drug
safety, 24(7), pp.709-715.
62. Pacurariu, A.C., Coloma, P.M., van Haren, A., Genov, G., Sturkenboom, M.C. and
Straus, S.M., 2014. A description of signals during the first 18 months of the EMA
pharmacovigilance risk assessment committee. Drug safety, 37(12), pp.1059-1066.
63. Vishal, P., Rahulgiri, G., Pratik, M. and Badjatya, J.K., 2014. A review on drug
approval process for US, Europe and India. International Journal of Drug Regulatory
Affairs, 2(1), pp.1-11.
64. Leyens, L. and Brand, A., 2016. Early patient access to medicines: health technology
assessment bodies need to catch up with new marketing authorization methods. Public
Health Genomics, 19(3), pp.187-191.
65. Garattini, L. and Padula, A., 2017. Why EMA should provide clearer guidance on the
authorization of NBCDs in generic and hybrid applications.
66. Monami, M., Dicembrini, I. and Mannucci, E., 2014. Dipeptidyl peptidase‐4
inhibitors and pancreatitis risk: a meta‐analysis of randomized clinical trials. Diabetes,
Obesity and Metabolism, 16(1), pp.48-56.
67. Krcevski‐Skvarc, N., Wells, C. and Häuser, W., 2018. Availability and approval of
cannabis‐based medicines for chronic pain management and palliative/supportive care
in Europe: A survey of the status in the chapters of the European Pain Federation.
European Journal of Pain, 22(3), pp.440-454.
52

68. Kuemmerle-Deschner, J.B., Ozen, S., Tyrrell, P.N., Kone-Paut, I., Goldbach-Mansky,
R., Lachmann, H., Blank, N., Hoffman, H.M., Weissbarth-Riedel, E., Hugle, B. and
Kallinich, T., 2017. Diagnostic criteria for cryopyrin-associated periodic syndrome
(CAPS). Annals of the rheumatic diseases, 76(6), pp.942-947.
69. Mullard, A., 2015. 2014 FDA drug approvals.
70. Ciociola, A.A., Cohen, L.B., Kulkarni, P., Kefalas, C., Buchman, A., Burke, C., Cain,
T., Connor, J., Ehrenpreis, E.D., Fang, J. and Fass, R., 2014. How drugs are
developed and approved by the FDA: current process and future directions. The
American journal of gastroenterology, 109(5), p.620.
71. Lee, D.A.D., 2014. The Hon. Michelle K. Lee Deputy Under Secretary of Commerce
Acting Directo Cremona, M., 2015. Guest editorial: negotiating the transatlantic trade
and investment partnership (TTIP). Common Market Law Review, 52(2), pp.351-362.
r United States Patent and Trademark Office.
72. Nikfarjam, A., Sarker, A., O’Connor, K., Ginn, R. and Gonzalez, G., 2015.
Pharmacovigilance from social media: mining adverse drug reaction mentions using
sequence labeling with word embedding cluster features. Journal of the American
Medical Informatics Association, 22(3), pp.671-681.
73. Prasad, V. and Mailankody, S., 2014. The accelerated approval of oncologic drugs:
lessons from ponatinib. JAMA, 311(4), pp.353-354.
74. Bertrand, M., Bombardini, M., Fisman, R., Hackinen, B. and Trebbi, F., 2018. Hall of
mirrors: Corporate philanthropy and strategic advocacy (No. w25329). National
Bureau of Economic Research.
75. Citeseerx.ist.psu.edu. (2019). [online] Available at:
http://citeseerx.ist.psu.edu/viewdoc/download?
doi=10.1.1.375.519&rep=rep1&type=pdf [Accessed 31 Jul. 2019].
53
R., Lachmann, H., Blank, N., Hoffman, H.M., Weissbarth-Riedel, E., Hugle, B. and
Kallinich, T., 2017. Diagnostic criteria for cryopyrin-associated periodic syndrome
(CAPS). Annals of the rheumatic diseases, 76(6), pp.942-947.
69. Mullard, A., 2015. 2014 FDA drug approvals.
70. Ciociola, A.A., Cohen, L.B., Kulkarni, P., Kefalas, C., Buchman, A., Burke, C., Cain,
T., Connor, J., Ehrenpreis, E.D., Fang, J. and Fass, R., 2014. How drugs are
developed and approved by the FDA: current process and future directions. The
American journal of gastroenterology, 109(5), p.620.
71. Lee, D.A.D., 2014. The Hon. Michelle K. Lee Deputy Under Secretary of Commerce
Acting Directo Cremona, M., 2015. Guest editorial: negotiating the transatlantic trade
and investment partnership (TTIP). Common Market Law Review, 52(2), pp.351-362.
r United States Patent and Trademark Office.
72. Nikfarjam, A., Sarker, A., O’Connor, K., Ginn, R. and Gonzalez, G., 2015.
Pharmacovigilance from social media: mining adverse drug reaction mentions using
sequence labeling with word embedding cluster features. Journal of the American
Medical Informatics Association, 22(3), pp.671-681.
73. Prasad, V. and Mailankody, S., 2014. The accelerated approval of oncologic drugs:
lessons from ponatinib. JAMA, 311(4), pp.353-354.
74. Bertrand, M., Bombardini, M., Fisman, R., Hackinen, B. and Trebbi, F., 2018. Hall of
mirrors: Corporate philanthropy and strategic advocacy (No. w25329). National
Bureau of Economic Research.
75. Citeseerx.ist.psu.edu. (2019). [online] Available at:
http://citeseerx.ist.psu.edu/viewdoc/download?
doi=10.1.1.375.519&rep=rep1&type=pdf [Accessed 31 Jul. 2019].
53

76. Ijpacr.com. (2019). [online] Available at: http://www.ijpacr.com/files/07-04-
2017/30.pdf [Accessed 31 Jul. 2019].
54
2017/30.pdf [Accessed 31 Jul. 2019].
54
1 out of 55
Related Documents






Your All-in-One AI-Powered Toolkit for Academic Success.
+13062052269
info@desklib.com
Available 24*7 on WhatsApp / Email
![[object Object]](/_next/static/media/star-bottom.7253800d.svg)
Unlock your academic potential
© 2024 | Zucol Services PVT LTD | All rights reserved.