Comprehensive Nursing Report: Tay-Sachs Disease, EPHLTH170
VerifiedAdded on 2023/03/20
|8
|2220
|21
Report
AI Summary
This nursing report comprehensively examines Tay-Sachs disease, an autosomal recessive genetic disorder prevalent among Ashkenazi Jews. The report is structured into three sections, each targeting a specific audience and purpose. Section 1 presents the disease's clinical manifestations, including motor deterioration, blindness, and cognitive decline, using formal medical terminology for an audience of doctors and nurses. It details the genetic basis of the disease, the deficiency of the Hexosaminidase A enzyme, and the diagnostic and treatment options available, emphasizing supportive care and seizure management. Section 2 translates this information for patients and their families, employing everyday language to describe the disease's inheritance pattern, symptoms, and progression. The report also explains the genetic inheritance patterns and disease progression in detail. Section 3 reflects on the learning experience, highlighting the importance of adapting communication styles for different audiences and proposing nursing interventions focused on genetic screening and patient education to prevent the disease's transmission. The report also includes references and an appendix with a glossary of medical terms.

Running head: NURSING
Nursing
Name of the Student
Name of the University
Author Note
Nursing
Name of the Student
Name of the University
Author Note
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

1
NURSING
Section 1
Tay-Sachs disease (TSD) is an autosomal recessive disease that falls under the
category of Ashkenazi Jews as this disease is common among the Eastern European Jews.
This is best known sphingolipidosis that affects approximately one out of 3600 population
from Ashkenazi Jewish ancestry. The affected infants are presented by 6 months of age with
symptoms like poor feeding, lethargy and floppiness. It is a progressive mental disorder some
of the prominent symptoms and signs of the disease include motor deterioration, blindness,
hepatosplenomegaly, bone lesions, pigmentation indifference to pain, emotional liability, lack
of tears and hyperhidrosis (Hussein et al., 2018). The loss of the developmental milestones or
developmental regression mainly becomes apparent during the second half of the first year of
disease development. As the feeding becomes increasingly difficulty, the infant progressively
deteriorates along with visible signs of visual impairment, deafness, spasticity those
progresses to rigidity. Death mainly occurs by the age of 3 due to respiratory infection
(Schneck & Volk, 2017).
The diagnosis of the TSD is supported clinically by the presence of a "cherry-red"
spot at the centre of the macula of fundus. The biochemical confirmatory test done for TSD is
demonstrated by decreased level of hexosaminidase A (Hex A) levels in the serum, or
cultured fibroblasts or white blood cells. Hex-A is mainly present in chromosome 15,
inheritance of the faulty gene, leads to manipulation of the gene in the chromosome 15
leading to generation of faulty Hex-A enzyme. Faulty Hex-Z enzyme has reduced activity.
The reduced activity of hexosaminidase A arises due to the deficiency of alpha subunit of the
enzyme beta-hexosaminidase that leads to the deposition of sphingolipid, GM2 ganglioside.
Deficiency of beta-subunit of the beta-hexosaminidase leads to the development of decreased
activity of the hexosaminidase B isozyme and thereby causing other GM2 gangliosidosis like
the Sandhoff diseases. The clinical presentation of TSD and Sandhoff diseases is identical
(Hussein et al., 2018).
The juvenile (subacute), chronic, and adult-onset variants of hexosaminidase A
deficiency generally have shows late onsets. The late onset of the disease is characterized by
slower disease progression. It is also associated with diverse neurological complications like
progressive dystonia, motor neuron disease and spinocerebellar degeneration. A bipolar form
NURSING
Section 1
Tay-Sachs disease (TSD) is an autosomal recessive disease that falls under the
category of Ashkenazi Jews as this disease is common among the Eastern European Jews.
This is best known sphingolipidosis that affects approximately one out of 3600 population
from Ashkenazi Jewish ancestry. The affected infants are presented by 6 months of age with
symptoms like poor feeding, lethargy and floppiness. It is a progressive mental disorder some
of the prominent symptoms and signs of the disease include motor deterioration, blindness,
hepatosplenomegaly, bone lesions, pigmentation indifference to pain, emotional liability, lack
of tears and hyperhidrosis (Hussein et al., 2018). The loss of the developmental milestones or
developmental regression mainly becomes apparent during the second half of the first year of
disease development. As the feeding becomes increasingly difficulty, the infant progressively
deteriorates along with visible signs of visual impairment, deafness, spasticity those
progresses to rigidity. Death mainly occurs by the age of 3 due to respiratory infection
(Schneck & Volk, 2017).
The diagnosis of the TSD is supported clinically by the presence of a "cherry-red"
spot at the centre of the macula of fundus. The biochemical confirmatory test done for TSD is
demonstrated by decreased level of hexosaminidase A (Hex A) levels in the serum, or
cultured fibroblasts or white blood cells. Hex-A is mainly present in chromosome 15,
inheritance of the faulty gene, leads to manipulation of the gene in the chromosome 15
leading to generation of faulty Hex-A enzyme. Faulty Hex-Z enzyme has reduced activity.
The reduced activity of hexosaminidase A arises due to the deficiency of alpha subunit of the
enzyme beta-hexosaminidase that leads to the deposition of sphingolipid, GM2 ganglioside.
Deficiency of beta-subunit of the beta-hexosaminidase leads to the development of decreased
activity of the hexosaminidase B isozyme and thereby causing other GM2 gangliosidosis like
the Sandhoff diseases. The clinical presentation of TSD and Sandhoff diseases is identical
(Hussein et al., 2018).
The juvenile (subacute), chronic, and adult-onset variants of hexosaminidase A
deficiency generally have shows late onsets. The late onset of the disease is characterized by
slower disease progression. It is also associated with diverse neurological complications like
progressive dystonia, motor neuron disease and spinocerebellar degeneration. A bipolar form

2
NURSING
of psychosis is highlighted in some individuals who have adult-onset of the disease (Schneck
& Volk, 2017).
Deik and Saunders‐Pullman (2014) stated that treatment for this genetic disorder. The
treatment options are available for the management of the signs and symptoms or clinical
manifestations of the disease. Treatment is mainly supportive and is directed towards
delivering proper nutrition, managing the water content of body and protecting the airway
constrictions or infection and effective control of seizures. The control of seizures is mainly
achieved by the use of conventional antiepileptic drugs like phenytoins, benzodiazepines,
phenytoins, and/or barbiturates. The dosage, route and time of administration of the drugs
vary with the type and severity of seizures. Conventional antipsychotic medications and
antidepressant therapy is used for the individuals with adult onset of hexosaminidase-A
deficiency along with psychiatric manifestations (U.S National Library of Medicine, 2019).
Recent pilot study conducted by Osher et al. (2015) was based on identification of the
effective treatment for the management of the late onset of TSD (LOTS). The authors mainly
choose Pyrimethamine (PMT) in order to modify the activity HexA. The results highlighted
that the cyclic dosage of PMT helps to increase HexA activity among the LOTS patients.
However, the observed increase is repeatedly transient and it is not directly associated with
the discernible beneficial neurological or other psychiatric effects.
According to Lew et al. (2015), the Australian Jewish community who reside in
Melbourde and Sydney are mainly affected by the disease. The estimated number of the
effected individual includes 97,300 during 2011 as per the Australian Bureau of Statistics
Census of Population and Housing. The majority of the Australian Jews are the carrier of the
TSD disease and the frequency is estimated to be approximately one among 25.
Section 2
TSD is a rare genetic disorder that is inherited in an autosomal recessive form. Such
that when both the parents (carrier) have one recessive gene for TSD (carrier), there is 25%
of change of giving birth to diseased child.
Rr (carrier parent) R (normal dominant gene) R (recessive gene)
Rr (carrier parent)
R RR (normal) Rr (carrier)
r rR (carrier) Rr (Diseased)
NURSING
of psychosis is highlighted in some individuals who have adult-onset of the disease (Schneck
& Volk, 2017).
Deik and Saunders‐Pullman (2014) stated that treatment for this genetic disorder. The
treatment options are available for the management of the signs and symptoms or clinical
manifestations of the disease. Treatment is mainly supportive and is directed towards
delivering proper nutrition, managing the water content of body and protecting the airway
constrictions or infection and effective control of seizures. The control of seizures is mainly
achieved by the use of conventional antiepileptic drugs like phenytoins, benzodiazepines,
phenytoins, and/or barbiturates. The dosage, route and time of administration of the drugs
vary with the type and severity of seizures. Conventional antipsychotic medications and
antidepressant therapy is used for the individuals with adult onset of hexosaminidase-A
deficiency along with psychiatric manifestations (U.S National Library of Medicine, 2019).
Recent pilot study conducted by Osher et al. (2015) was based on identification of the
effective treatment for the management of the late onset of TSD (LOTS). The authors mainly
choose Pyrimethamine (PMT) in order to modify the activity HexA. The results highlighted
that the cyclic dosage of PMT helps to increase HexA activity among the LOTS patients.
However, the observed increase is repeatedly transient and it is not directly associated with
the discernible beneficial neurological or other psychiatric effects.
According to Lew et al. (2015), the Australian Jewish community who reside in
Melbourde and Sydney are mainly affected by the disease. The estimated number of the
effected individual includes 97,300 during 2011 as per the Australian Bureau of Statistics
Census of Population and Housing. The majority of the Australian Jews are the carrier of the
TSD disease and the frequency is estimated to be approximately one among 25.
Section 2
TSD is a rare genetic disorder that is inherited in an autosomal recessive form. Such
that when both the parents (carrier) have one recessive gene for TSD (carrier), there is 25%
of change of giving birth to diseased child.
Rr (carrier parent) R (normal dominant gene) R (recessive gene)
Rr (carrier parent)
R RR (normal) Rr (carrier)
r rR (carrier) Rr (Diseased)
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

3
NURSING
RR (normal) 25%
Rr (carrier) 50%
Rr (Diseased) 25%
Table: Inheritance Pattern on TSD
Source: Created by Author
The disease is fatal that caused progressive destruction of the nervous system. It is
mainly caused due to the absence of a vital enzyme of the body known as hexosaminidase A
(Hex A). Hex-A gene is located in the chromosome 15, inherence of both the copies of the
faulty gene in leads to the generation of truncated protein (broken protein) and hampers the
activity of the Hex-A enzyme. In the absence of the enzyme Hex-A, a fatty acid substance or
lipid, known as GM2 ganglioside accumulates within the cells. It mainly targets the neurons
of the brain. This progressive accumulation of GM2 ganglioside in brain cells or neurons of
the brain leads to damage of the brain cells. The destruction of the neurons leads to the
generation of several neurological complications.
In other others it can be said that every individual have two copies of Hex-A gene. If
either one or both the Hex-A copies of gene is active, the body is capable of producing
adequate enzyme in order to prevent the abnormal build-up of the GM2 ganglioside lipid in
the neurons. Carriers of TSD have one copy of inactive gene and one copy of active gene and
are healthy but they are capable to passing on the disease to a child.
In the new born child, the destructive process mainly initiates when the child is in
fetus that is during the early stage of pregnancy. However, during the birth, the baby appears
normal until six months of age the disease slowly manifests its symptoms. By two years of
age, the affected children experiences symptoms like recurrent seizures along with decreased
level of mental function. The child gradually regressed and is even unable to move by
crawling, or conduct other mobility activities like turning over the bed, sit over the bed or
reaching out of any objects. Gradually the child become blind and looses the hearing
capability (deaf) and the entire cognitive system becomes paralysed or non-responsive. By
the time the child is three to four years only, the central nervous system of the body is
NURSING
RR (normal) 25%
Rr (carrier) 50%
Rr (Diseased) 25%
Table: Inheritance Pattern on TSD
Source: Created by Author
The disease is fatal that caused progressive destruction of the nervous system. It is
mainly caused due to the absence of a vital enzyme of the body known as hexosaminidase A
(Hex A). Hex-A gene is located in the chromosome 15, inherence of both the copies of the
faulty gene in leads to the generation of truncated protein (broken protein) and hampers the
activity of the Hex-A enzyme. In the absence of the enzyme Hex-A, a fatty acid substance or
lipid, known as GM2 ganglioside accumulates within the cells. It mainly targets the neurons
of the brain. This progressive accumulation of GM2 ganglioside in brain cells or neurons of
the brain leads to damage of the brain cells. The destruction of the neurons leads to the
generation of several neurological complications.
In other others it can be said that every individual have two copies of Hex-A gene. If
either one or both the Hex-A copies of gene is active, the body is capable of producing
adequate enzyme in order to prevent the abnormal build-up of the GM2 ganglioside lipid in
the neurons. Carriers of TSD have one copy of inactive gene and one copy of active gene and
are healthy but they are capable to passing on the disease to a child.
In the new born child, the destructive process mainly initiates when the child is in
fetus that is during the early stage of pregnancy. However, during the birth, the baby appears
normal until six months of age the disease slowly manifests its symptoms. By two years of
age, the affected children experiences symptoms like recurrent seizures along with decreased
level of mental function. The child gradually regressed and is even unable to move by
crawling, or conduct other mobility activities like turning over the bed, sit over the bed or
reaching out of any objects. Gradually the child become blind and looses the hearing
capability (deaf) and the entire cognitive system becomes paralysed or non-responsive. By
the time the child is three to four years only, the central nervous system of the body is
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

4
NURSING
disrupted completely and this eventually leads to death. The maximum age span of TSD
infected child is 5 years.
One of the abnormalities that is helpful in the identification of the disease is presence
of cherry-red spot over the eyes and is detected through eye examination. The symptoms that
appear during the adult onset of the disease are milder in comparison to the infantile or early
onset of the disease.
The disease is mainly common among the eastern European Ashkenazi Jewsign
population. There is no cure or effective treatment of TASD. However, the scientists are at
present exploring the enzymatic therapy for the treatment of the disease. At presented bone
marrow transplantation is explored in order to reverse the disease progression or slowing the
damage of the central nervous systems of the babies.
Section 3
The detailed study about the TSD helped me in understanding the mode of
transformation of the genetic disease. It helped me to understand that carriers of the
autosomal recessive disorder are normal with normal psychological and biochemical
functioning. However, they have the capability to transferring the faulty gene or the faulty
recessive gene in their child. When both the child receives the faulty gene (in this case both
the parents are required to be carriers), the child become diseases. The disease progression of
TSD also showed that how improper functioning of a single enzyme lead to the development
of fatal outcomes. It helped me to understand the each and every enzyme secreted in the body
have an important function and inheritance of the faulty gene hamper the normal functioning
of the enzyme.
The section, which I found extremely interesting while writing are: the signs and
symptoms of the disease. This section was easier also because it had less complicated terms
that are easy to understand. Moreover, while writing the section two, I understood that how
the dialect and the nature of the communication style changes. For example while writing
disease information for the medical professionals, I used scientific terms and the same
information when I was writing from a lay man, I used simpler and easy to understand
language and thus helping me to get a detailed understanding of change in communication
while handling patients during informed decision making.
NURSING
disrupted completely and this eventually leads to death. The maximum age span of TSD
infected child is 5 years.
One of the abnormalities that is helpful in the identification of the disease is presence
of cherry-red spot over the eyes and is detected through eye examination. The symptoms that
appear during the adult onset of the disease are milder in comparison to the infantile or early
onset of the disease.
The disease is mainly common among the eastern European Ashkenazi Jewsign
population. There is no cure or effective treatment of TASD. However, the scientists are at
present exploring the enzymatic therapy for the treatment of the disease. At presented bone
marrow transplantation is explored in order to reverse the disease progression or slowing the
damage of the central nervous systems of the babies.
Section 3
The detailed study about the TSD helped me in understanding the mode of
transformation of the genetic disease. It helped me to understand that carriers of the
autosomal recessive disorder are normal with normal psychological and biochemical
functioning. However, they have the capability to transferring the faulty gene or the faulty
recessive gene in their child. When both the child receives the faulty gene (in this case both
the parents are required to be carriers), the child become diseases. The disease progression of
TSD also showed that how improper functioning of a single enzyme lead to the development
of fatal outcomes. It helped me to understand the each and every enzyme secreted in the body
have an important function and inheritance of the faulty gene hamper the normal functioning
of the enzyme.
The section, which I found extremely interesting while writing are: the signs and
symptoms of the disease. This section was easier also because it had less complicated terms
that are easy to understand. Moreover, while writing the section two, I understood that how
the dialect and the nature of the communication style changes. For example while writing
disease information for the medical professionals, I used scientific terms and the same
information when I was writing from a lay man, I used simpler and easy to understand
language and thus helping me to get a detailed understanding of change in communication
while handling patients during informed decision making.

5
NURSING
From the perspective of the nursing intervention, I would like to highlight the since
the disease has no cure, I would try to eradicate the disease from the grass route level. This
can be done by educating the couples of this genetic disease and who genetic screening like
the one done in case of thalassemia can be effective in controlling the birth of diseases child.
NURSING
From the perspective of the nursing intervention, I would like to highlight the since
the disease has no cure, I would try to eradicate the disease from the grass route level. This
can be done by educating the couples of this genetic disease and who genetic screening like
the one done in case of thalassemia can be effective in controlling the birth of diseases child.
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

6
NURSING
References
Deik, A., & Saunders‐Pullman, R. (2014). Atypical presentation of late‐onset Tay‐sachs
disease. Muscle & nerve, 49(5), 768-771. https://doi.org/10.1002/mus.24146
Hussein, N., Weng, S. F., Kai, J., Kleijnen, J., & Qureshi, N. (2018). Preconception risk
assessment for thalassaemia, sickle cell disease, cystic fibrosis and Tay‐Sachs
disease. Cochrane Database of Systematic Reviews, (3). Retrieved from:
https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD010849.pub3/
abstract
Lew, R. M., Burnett, L., Proos, A. L., & Delatycki, M. B. (2015). Tay-Sachs disease: current
perspectives from Australia. The application of clinical genetics, 8, 19.
doi: 10.2147/TACG.S49628
Osher, E., Fattal-Valevski, A., Sagie, L., Urshanski, N., Sagiv, N., Peleg, L., ... & Valevski,
A. (2015). Effect of cyclic, low dose pyrimethamine treatment in patients with late
onset Tay Sachs: an open label, extended pilot study. Orphanet journal of rare
diseases, 10(1), 45. https://doi.org/10.1186/s13023-015-0260-7
Schneck, l., & Volk, B. W. (2017, January). Clinical manifestations of Tay-Sachs disease and
Niemann-Pick disease. In Inborn Disorders of Sphingolipid Metabolism: Proceedings
of the Third International Symposium on the Cerebral Sphingolipidoses (p. 403).
Elsevier. https://doi.org/10.1186/s13023-015-0260-7
U.S National Library of Medicine. (2019). Tay-Sachs disease Access date: 11th May 2019.
Retrieved from: https://ghr.nlm.nih.gov/condition/tay-sachs-disease
NURSING
References
Deik, A., & Saunders‐Pullman, R. (2014). Atypical presentation of late‐onset Tay‐sachs
disease. Muscle & nerve, 49(5), 768-771. https://doi.org/10.1002/mus.24146
Hussein, N., Weng, S. F., Kai, J., Kleijnen, J., & Qureshi, N. (2018). Preconception risk
assessment for thalassaemia, sickle cell disease, cystic fibrosis and Tay‐Sachs
disease. Cochrane Database of Systematic Reviews, (3). Retrieved from:
https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD010849.pub3/
abstract
Lew, R. M., Burnett, L., Proos, A. L., & Delatycki, M. B. (2015). Tay-Sachs disease: current
perspectives from Australia. The application of clinical genetics, 8, 19.
doi: 10.2147/TACG.S49628
Osher, E., Fattal-Valevski, A., Sagie, L., Urshanski, N., Sagiv, N., Peleg, L., ... & Valevski,
A. (2015). Effect of cyclic, low dose pyrimethamine treatment in patients with late
onset Tay Sachs: an open label, extended pilot study. Orphanet journal of rare
diseases, 10(1), 45. https://doi.org/10.1186/s13023-015-0260-7
Schneck, l., & Volk, B. W. (2017, January). Clinical manifestations of Tay-Sachs disease and
Niemann-Pick disease. In Inborn Disorders of Sphingolipid Metabolism: Proceedings
of the Third International Symposium on the Cerebral Sphingolipidoses (p. 403).
Elsevier. https://doi.org/10.1186/s13023-015-0260-7
U.S National Library of Medicine. (2019). Tay-Sachs disease Access date: 11th May 2019.
Retrieved from: https://ghr.nlm.nih.gov/condition/tay-sachs-disease
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser
7
NURSING
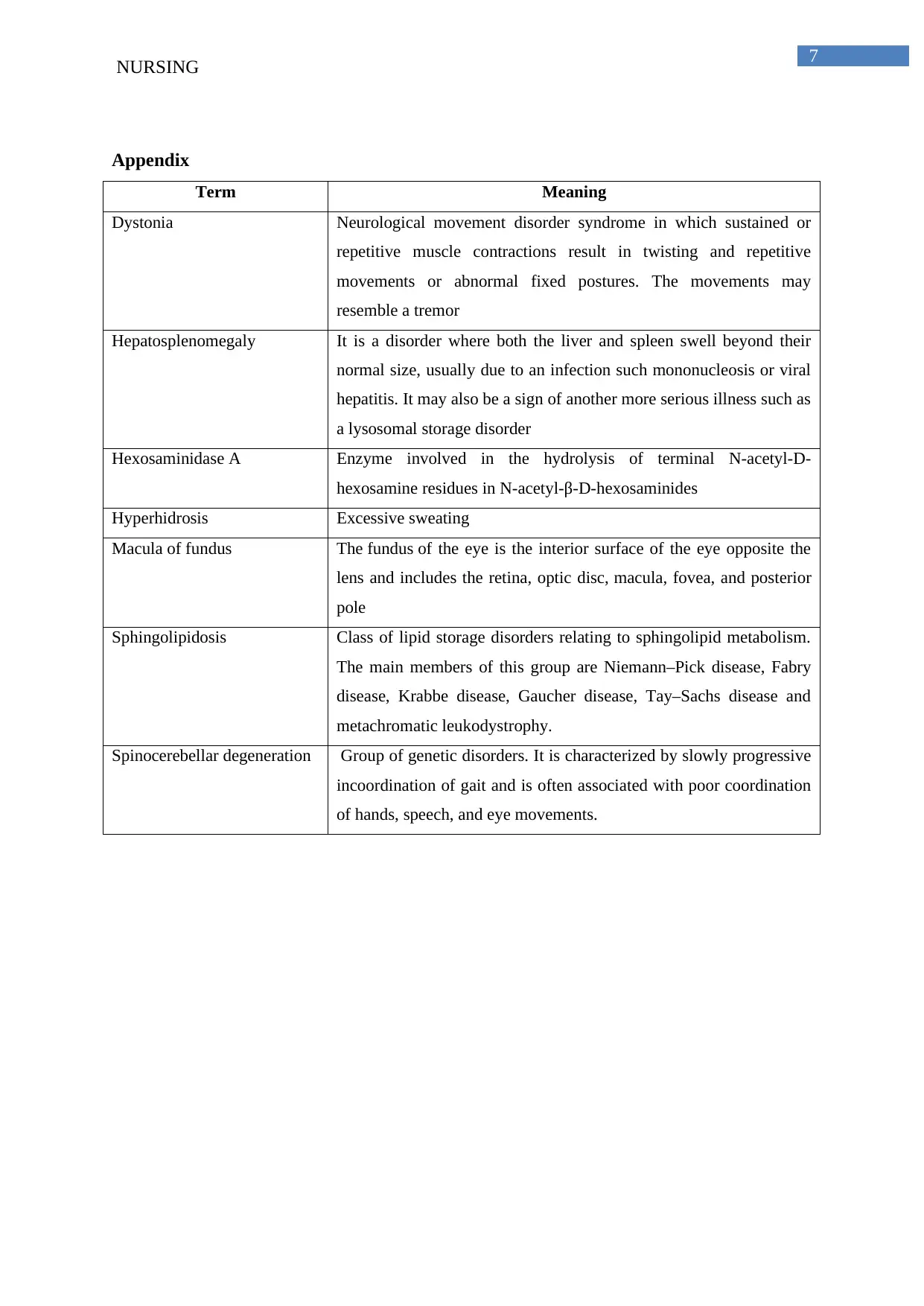
Appendix
Term Meaning
Dystonia Neurological movement disorder syndrome in which sustained or
repetitive muscle contractions result in twisting and repetitive
movements or abnormal fixed postures. The movements may
resemble a tremor
Hepatosplenomegaly It is a disorder where both the liver and spleen swell beyond their
normal size, usually due to an infection such mononucleosis or viral
hepatitis. It may also be a sign of another more serious illness such as
a lysosomal storage disorder
Hexosaminidase A Enzyme involved in the hydrolysis of terminal N-acetyl-D-
hexosamine residues in N-acetyl-β-D-hexosaminides
Hyperhidrosis Excessive sweating
Macula of fundus The fundus of the eye is the interior surface of the eye opposite the
lens and includes the retina, optic disc, macula, fovea, and posterior
pole
Sphingolipidosis Class of lipid storage disorders relating to sphingolipid metabolism.
The main members of this group are Niemann–Pick disease, Fabry
disease, Krabbe disease, Gaucher disease, Tay–Sachs disease and
metachromatic leukodystrophy.
Spinocerebellar degeneration Group of genetic disorders. It is characterized by slowly progressive
incoordination of gait and is often associated with poor coordination
of hands, speech, and eye movements.
NURSING
Appendix
Term Meaning
Dystonia Neurological movement disorder syndrome in which sustained or
repetitive muscle contractions result in twisting and repetitive
movements or abnormal fixed postures. The movements may
resemble a tremor
Hepatosplenomegaly It is a disorder where both the liver and spleen swell beyond their
normal size, usually due to an infection such mononucleosis or viral
hepatitis. It may also be a sign of another more serious illness such as
a lysosomal storage disorder
Hexosaminidase A Enzyme involved in the hydrolysis of terminal N-acetyl-D-
hexosamine residues in N-acetyl-β-D-hexosaminides
Hyperhidrosis Excessive sweating
Macula of fundus The fundus of the eye is the interior surface of the eye opposite the
lens and includes the retina, optic disc, macula, fovea, and posterior
pole
Sphingolipidosis Class of lipid storage disorders relating to sphingolipid metabolism.
The main members of this group are Niemann–Pick disease, Fabry
disease, Krabbe disease, Gaucher disease, Tay–Sachs disease and
metachromatic leukodystrophy.
Spinocerebellar degeneration Group of genetic disorders. It is characterized by slowly progressive
incoordination of gait and is often associated with poor coordination
of hands, speech, and eye movements.
1 out of 8
Your All-in-One AI-Powered Toolkit for Academic Success.
+13062052269
info@desklib.com
Available 24*7 on WhatsApp / Email
![[object Object]](/_next/static/media/star-bottom.7253800d.svg)
Unlock your academic potential
Copyright © 2020–2026 A2Z Services. All Rights Reserved. Developed and managed by ZUCOL.