Exploring Louis' Oculomotor Apraxia and DD Through Genetic Analysis
VerifiedAdded on 2023/04/21
|17
|6149
|494
Case Study
AI Summary
This case study delves into the genetic analysis of Louis, a 15-year-old male affected by mild to moderate intellectual disability, developmental delay, and oculomotor apraxia. It critically analyzes Louis's family history, including his father's pancreatic carcinoma and his niece's polydactyly, to assess potential genetic risks. The study explores the appropriateness of genome sequencing in identifying pathogenic allelic variations in genes like APTX, PIK3R5, PNKP, and SETX, to rule out differential diagnoses and understand the inheritance pattern of Louis's condition. Furthermore, the case study assesses three reported genetic variants related to CEP290, C5ORF42, and BRCA2 genes, examining their molecular biological consequences, allele frequencies, and likely pathogenicity in relation to Louis's phenotypes. The analysis also touches upon the ethical implications for family members and potential further testing or treatment implications for Louis and his family. Desklib provides similar solved assignments and resources for students.

Page 1 of 17
Table of Content
Part – A………………………………………………………………………………………………………………………………………………2
Key Information for History Taking – Critical Analysis……………………………………………………………2
Pedigree (Family Tree) ………………………………………………………………………………………….………………4
Likely Mode of Inheritance of Louis' Condition…………………………………………………….……………….5
Why Genome Sequencing Might be Appropriate in the Present Situation? – A Critical
Analysis…………………………………………………………………………………………………………………………………6
Part – B…………………………………………………………………………………………………………………………….……………….7
Assessment of the Reported Three Variants. ………………………………………………………………..……..7
Molecular Biological Consequence………………………………………………………………………………………..7
Allele Frequency.. …………………………………………………………………………………………………………………8
Likely Pathogenicity………………………………………………………………………………………………………………9
Diagnoses and their Relevance to Louis' Phenotypes………………………………………………………….10
Ethical Implications for Family Members………………………………………………………………………….…11
Further Testing or Treatment Implications for Louis and Other Family Members……………….12
References………………………………………………………………………………………………………………………....13
Table of Content
Part – A………………………………………………………………………………………………………………………………………………2
Key Information for History Taking – Critical Analysis……………………………………………………………2
Pedigree (Family Tree) ………………………………………………………………………………………….………………4
Likely Mode of Inheritance of Louis' Condition…………………………………………………….……………….5
Why Genome Sequencing Might be Appropriate in the Present Situation? – A Critical
Analysis…………………………………………………………………………………………………………………………………6
Part – B…………………………………………………………………………………………………………………………….……………….7
Assessment of the Reported Three Variants. ………………………………………………………………..……..7
Molecular Biological Consequence………………………………………………………………………………………..7
Allele Frequency.. …………………………………………………………………………………………………………………8
Likely Pathogenicity………………………………………………………………………………………………………………9
Diagnoses and their Relevance to Louis' Phenotypes………………………………………………………….10
Ethical Implications for Family Members………………………………………………………………………….…11
Further Testing or Treatment Implications for Louis and Other Family Members……………….12
References………………………………………………………………………………………………………………………....13
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

Page 2 of 17
Part – A
Key Information for History Taking – Critical Analysis
The key information presented in the scenario relates to the following attributes.
Louis (i.e. the male patient) is affected with intellectual disability (ID) pertaining to a
mild/moderate level as well as developmental delay (DD). These disorders are based on
phenotypic disruptions that lead to the development of psychological challenges and
psychosomatic complications in the affected children. The genetic assessment of DD/ID is
based on the diagnostic interventions including mitochondrial DNA testing, gene panel, a
single gene analysis, Fragile X assessment, microarray, and karyotype (Bowling et al. 2017).
However, the whole genome or exome sequencing is based on the comprehensive diagnostic
assay that provides significant information on the concerned genomic variants through their
high-resolution assessment. Genetic variants of DD/ID include benign, likely benign, VUS,
likely pathogenic, and pathogenic types. Genetic variations in children affected with DD/ID
lead to the development of nonsense, frameshift, or missense effects. Genetic studies of the
significant variants of DD/ID reveal substantial variations in the transcription factor EBF3
that impact the overall neurodevelopment of the affected child. The EBF3-based deterioration
of the neurodevelopmental phenotypes is the most probable cause of intellectual
disability/developmental delay in the pediatric population. Developmental delay based on
sensory or motor impairments is diagnosed by assessing aneuploidy or polyploidy through
the utilization of G-banded karyotyping (Sun, Oristaglio & Levy 2015).
Louis is also a victim of oculomotor apraxia in the presented scenario. Oculomotor
apraxia in children is accompanied with ataxia, dysarthria, and peripheral neuropathy.
Children affected with oculomotor apraxia have atrophic and short feet and hands. The
disease progresses with mild/moderate cognitive impairment in most pediatric scenarios.
AOA1 (Ataxia with oculomotor apraxia type 1) is an autosomal recessive disorder that
requires the sequence assessment of APTX gene to evaluate its pathogenic variants (Adam,
Ardinger & Pagon 2019). Copy number/ or duplication/deletion assessment is the alternative
genetic test for evaluating AOA1 in the suspected children. AOA2 is also an autosomal
recessive disorder that causes cerebellar ataxia in the affected children (Adam, Ardinger &
Pagon n.d.). The sequence assessment of SETX gene assists in evaluating its splice site,
nonsense, and missense variants. This disease is not associated with whole-genes or exon
duplications/deletions. However, the gene-targeted assessment of SETX duplication/deletion
helps in identifying 8%-20% of the pathogenic variants. The occurrence of SETX gene in the
family increases the risk of oculomotor apraxia in the related children. Therefore, carrier
Part – A
Key Information for History Taking – Critical Analysis
The key information presented in the scenario relates to the following attributes.
Louis (i.e. the male patient) is affected with intellectual disability (ID) pertaining to a
mild/moderate level as well as developmental delay (DD). These disorders are based on
phenotypic disruptions that lead to the development of psychological challenges and
psychosomatic complications in the affected children. The genetic assessment of DD/ID is
based on the diagnostic interventions including mitochondrial DNA testing, gene panel, a
single gene analysis, Fragile X assessment, microarray, and karyotype (Bowling et al. 2017).
However, the whole genome or exome sequencing is based on the comprehensive diagnostic
assay that provides significant information on the concerned genomic variants through their
high-resolution assessment. Genetic variants of DD/ID include benign, likely benign, VUS,
likely pathogenic, and pathogenic types. Genetic variations in children affected with DD/ID
lead to the development of nonsense, frameshift, or missense effects. Genetic studies of the
significant variants of DD/ID reveal substantial variations in the transcription factor EBF3
that impact the overall neurodevelopment of the affected child. The EBF3-based deterioration
of the neurodevelopmental phenotypes is the most probable cause of intellectual
disability/developmental delay in the pediatric population. Developmental delay based on
sensory or motor impairments is diagnosed by assessing aneuploidy or polyploidy through
the utilization of G-banded karyotyping (Sun, Oristaglio & Levy 2015).
Louis is also a victim of oculomotor apraxia in the presented scenario. Oculomotor
apraxia in children is accompanied with ataxia, dysarthria, and peripheral neuropathy.
Children affected with oculomotor apraxia have atrophic and short feet and hands. The
disease progresses with mild/moderate cognitive impairment in most pediatric scenarios.
AOA1 (Ataxia with oculomotor apraxia type 1) is an autosomal recessive disorder that
requires the sequence assessment of APTX gene to evaluate its pathogenic variants (Adam,
Ardinger & Pagon 2019). Copy number/ or duplication/deletion assessment is the alternative
genetic test for evaluating AOA1 in the suspected children. AOA2 is also an autosomal
recessive disorder that causes cerebellar ataxia in the affected children (Adam, Ardinger &
Pagon n.d.). The sequence assessment of SETX gene assists in evaluating its splice site,
nonsense, and missense variants. This disease is not associated with whole-genes or exon
duplications/deletions. However, the gene-targeted assessment of SETX duplication/deletion
helps in identifying 8%-20% of the pathogenic variants. The occurrence of SETX gene in the
family increases the risk of oculomotor apraxia in the related children. Therefore, carrier

Page 3 of 17
testing is the method of choice to evaluate the genetic risk of reproductive organs and
offspring.
The presented scenario describes the demise of Louis’s father from pancreatic
carcinoma. Louis’s family history of pancreatic cancer clearly reveals his risk of developing
familial pancreatic cancer at a later stage in life. The significant genes that elevate the risk of
carcinoma include CDKN2A and BRCA2, ATM, and PALB2 (Petersen 2016). The germline
mutations in any of these genes become the cause of pancreatic cancer and its clinical
complications in the affected individuals. The genes including DNA mismatch repair genes,
STK11, SPINK1, PRSS1, PALB2, BRCA2 are regarded as the high-penetrance genes;
however, ABO and CFTR are considered as low penetrance genes for pancreatic cancer
(Klein 2012). The sequence assessment of these genes is therefore required in Louis’s case to
determine the genetic variants in the context of evaluating his risk of pancreatic cancer.
Louis’s youngest niece of age 6-months is affected with hands’ polydactyly. The
congenital limb malformation (i.e. polydactyly) is based on genetic mutations that directly
impact the ZPA (zone of polarizing activity) that controls the positional identity and limb
structure of individuals (Ahmed et al. 2017). The classification of polydactyly is based on
three distinct phenotypes, including postaxial, central, and preaxial. These conditions occur
under the impact of transcription factors that include GLI3, SHH, PAPA3, and PAPA2 across
chr7p13, chr7q36, chr19p13.2-p13.1, and chr13q21-q32 respectively. Furthermore,
polydactyly also relates to PITXI, MIPOLI, and SHH. Polydactyly is subjected to
reconstructive interventions to restore the normal hand anatomy (Comer, Potter & Ladd
2018).
testing is the method of choice to evaluate the genetic risk of reproductive organs and
offspring.
The presented scenario describes the demise of Louis’s father from pancreatic
carcinoma. Louis’s family history of pancreatic cancer clearly reveals his risk of developing
familial pancreatic cancer at a later stage in life. The significant genes that elevate the risk of
carcinoma include CDKN2A and BRCA2, ATM, and PALB2 (Petersen 2016). The germline
mutations in any of these genes become the cause of pancreatic cancer and its clinical
complications in the affected individuals. The genes including DNA mismatch repair genes,
STK11, SPINK1, PRSS1, PALB2, BRCA2 are regarded as the high-penetrance genes;
however, ABO and CFTR are considered as low penetrance genes for pancreatic cancer
(Klein 2012). The sequence assessment of these genes is therefore required in Louis’s case to
determine the genetic variants in the context of evaluating his risk of pancreatic cancer.
Louis’s youngest niece of age 6-months is affected with hands’ polydactyly. The
congenital limb malformation (i.e. polydactyly) is based on genetic mutations that directly
impact the ZPA (zone of polarizing activity) that controls the positional identity and limb
structure of individuals (Ahmed et al. 2017). The classification of polydactyly is based on
three distinct phenotypes, including postaxial, central, and preaxial. These conditions occur
under the impact of transcription factors that include GLI3, SHH, PAPA3, and PAPA2 across
chr7p13, chr7q36, chr19p13.2-p13.1, and chr13q21-q32 respectively. Furthermore,
polydactyly also relates to PITXI, MIPOLI, and SHH. Polydactyly is subjected to
reconstructive interventions to restore the normal hand anatomy (Comer, Potter & Ladd
2018).
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide
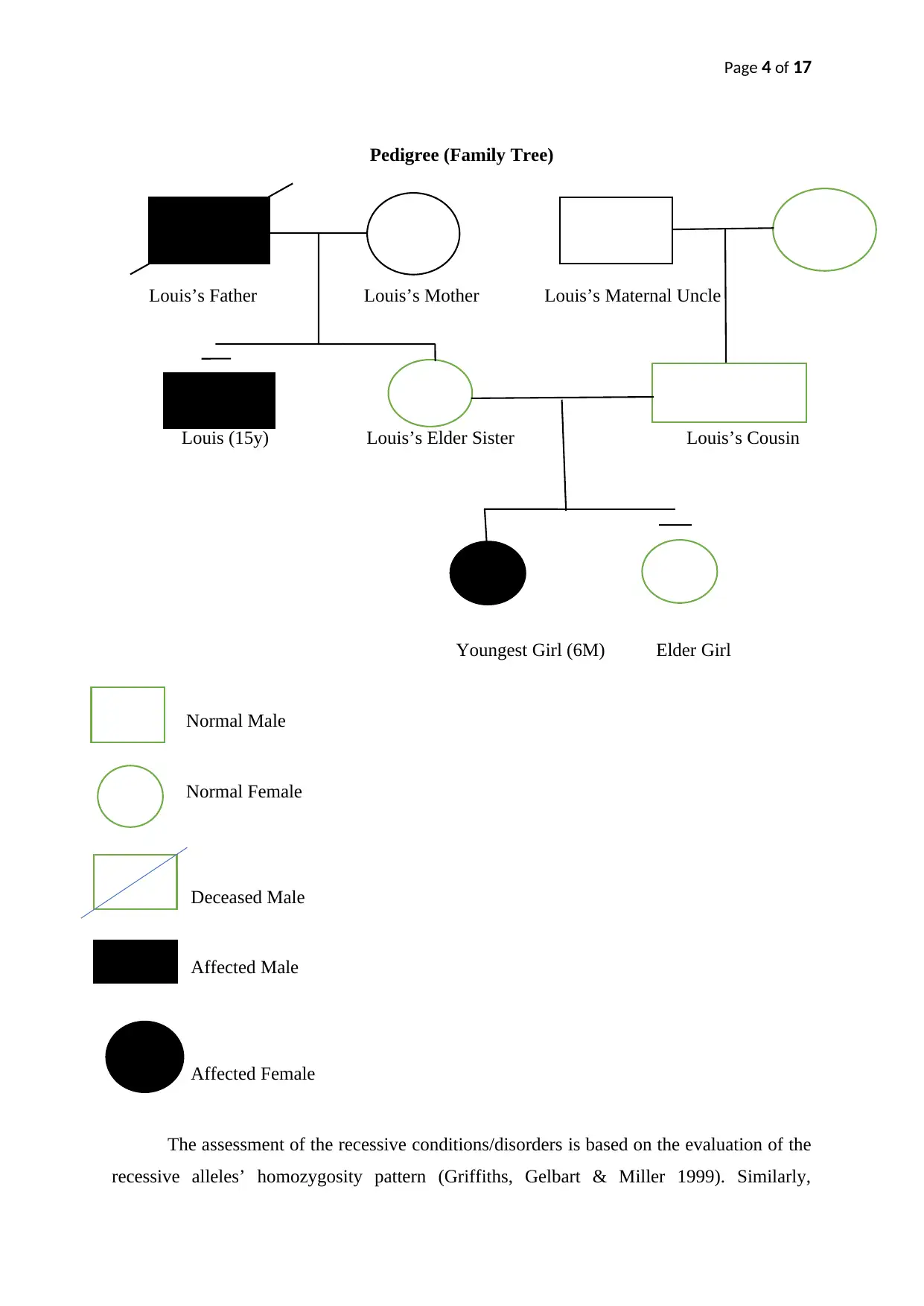
Page 4 of 17
Pedigree (Family Tree)
Louis’s Father Louis’s Mother Louis’s Maternal Uncle
Louis (15y) Louis’s Elder Sister Louis’s Cousin
Youngest Girl (6M) Elder Girl
Normal Male
Normal Female
Deceased Male
Affected Male
Affected Female
The assessment of the recessive conditions/disorders is based on the evaluation of the
recessive alleles’ homozygosity pattern (Griffiths, Gelbart & Miller 1999). Similarly,
Pedigree (Family Tree)
Louis’s Father Louis’s Mother Louis’s Maternal Uncle
Louis (15y) Louis’s Elder Sister Louis’s Cousin
Youngest Girl (6M) Elder Girl
Normal Male
Normal Female
Deceased Male
Affected Male
Affected Female
The assessment of the recessive conditions/disorders is based on the evaluation of the
recessive alleles’ homozygosity pattern (Griffiths, Gelbart & Miller 1999). Similarly,
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

Page 5 of 17
conditions including oculomotor apraxia with ataxia and developmental/intellectual delay are
also associated with autosomal recessive genes. That’s why oculomotor apraxia is regarded
as an autosomal recessive disorder (Németh et al. 2000). The dominant alleles give an
impression of the unaffected phenotype. The recessive allele also indicates the autosomal
inheritance pattern for the disease pattern (marked by the shaded exceptional phenotype (i.e.
the phenotypes of Louis, his father, and the youngest niece). Louis’s pedigree indicates the
heterozygous status of his parents who contributed the dominant and recessive alleles to their
children (including Louis and his sister). Since Louis father and mother were never affected
with oculomotor apraxia and intellectual disability, both could have contributed a dominant
normal ‘P’ allele along with the recessive diseased ‘p’ allele (pertaining to oculomotor
apraxia) whose different expression patterns in Louis resulted in his existing disease pattern.
Louis’s case could have exhibited the monohybrid cross and the resultant 1:3 ratio might be
responsible for the accumulation of both recessive alleles in Louis. This combination could
have contributed to the recessive phenotype expression in Louis that potentially induced the
development of oculomotor apraxia and associated clinical manifestations. The pictorial
representation of the monohybrid cross for the autosomal recessive disorder (i.e. oculomotor
with intellectual disability) is provided below.
1:3 Ratio
P p
P PP Pp
p Pp pp
P = Dominant Normal Allele
p = Recessive allele representing oculomotor apraxia
pp = Louis’s phenotype
Louis’s intrafamilial variability is responsible for his developmental delay and
oculomotor apraxia. This subtle variation leading to missense change and SETX variants
beyond the helicase domain indicate the abnormal functional activation of senataxin’s N-
terminus under the impact of the recessive phenotypic expression (Criscuolo et al. 2006).
Louis’s family tree categorically describes this inheritance pattern of the genetic variant that
resulted in the development of his congenital anomaly (i.e. oculomotor apraxia with
intellectual disability).
Likely Mode of Inheritance of Louis' Condition
conditions including oculomotor apraxia with ataxia and developmental/intellectual delay are
also associated with autosomal recessive genes. That’s why oculomotor apraxia is regarded
as an autosomal recessive disorder (Németh et al. 2000). The dominant alleles give an
impression of the unaffected phenotype. The recessive allele also indicates the autosomal
inheritance pattern for the disease pattern (marked by the shaded exceptional phenotype (i.e.
the phenotypes of Louis, his father, and the youngest niece). Louis’s pedigree indicates the
heterozygous status of his parents who contributed the dominant and recessive alleles to their
children (including Louis and his sister). Since Louis father and mother were never affected
with oculomotor apraxia and intellectual disability, both could have contributed a dominant
normal ‘P’ allele along with the recessive diseased ‘p’ allele (pertaining to oculomotor
apraxia) whose different expression patterns in Louis resulted in his existing disease pattern.
Louis’s case could have exhibited the monohybrid cross and the resultant 1:3 ratio might be
responsible for the accumulation of both recessive alleles in Louis. This combination could
have contributed to the recessive phenotype expression in Louis that potentially induced the
development of oculomotor apraxia and associated clinical manifestations. The pictorial
representation of the monohybrid cross for the autosomal recessive disorder (i.e. oculomotor
with intellectual disability) is provided below.
1:3 Ratio
P p
P PP Pp
p Pp pp
P = Dominant Normal Allele
p = Recessive allele representing oculomotor apraxia
pp = Louis’s phenotype
Louis’s intrafamilial variability is responsible for his developmental delay and
oculomotor apraxia. This subtle variation leading to missense change and SETX variants
beyond the helicase domain indicate the abnormal functional activation of senataxin’s N-
terminus under the impact of the recessive phenotypic expression (Criscuolo et al. 2006).
Louis’s family tree categorically describes this inheritance pattern of the genetic variant that
resulted in the development of his congenital anomaly (i.e. oculomotor apraxia with
intellectual disability).
Likely Mode of Inheritance of Louis' Condition

Page 6 of 17
Louis’s oculomotor apraxia along with developmental delay was inherited in an
autosomal recessive format (GHR 2019). Both mutated recessive genes were transferred from
parents to Louis in a manner that his cells contained both mutated genes copy whose overt
expression resulted in the development of oculomotor apraxia. Louis’s parents probably
carried the single version of the mutated recessive gene and that’s why never expressed the
clinical manifestations of oculomotor apraxia with developmental delay. Oculomotor apraxia
in the presented context exhibits 25% risk of its expression in an autosomal recessive pattern
(Coutinho 2019). This indicates that Louis and his sister has a 25% chance of acquiring this
congenital anomaly at their birth. Furthermore, they had a 25% chance of neither acquiring
the disease nor exhibiting the carrier status. Louis and his sister had a 50% probability of
becoming the symptomatic carriers. Louis in the presented scenario qualified the 25%
probability of acquiring the disease and eventually expressed the autosomal recessive pattern
of oculomotor apraxia. The homozygous mutation pattern that arrived from the phenotype
correlations in Louis’s case made him the victim of oculomotor apraxia. Furthermore, the AT
(ataxia) genes based on their compound heterozygosity in the could have promoted the
development of Louis’s AOA (ataxia with oculomotor apraxia) at birth (McKusick & Hartz
2017).
Why Genome Sequencing Might be Appropriate in the Present Situation? – A Critical
Analysis
DNA sequencing assists in tracking the nucleotide sequences in the genetic codes of
individuals. This technique is highly advantageous in terms of evaluating the mechanism of
genetic disorders. Whole genome sequencing assists in determining the genetic variants by
rapidly evaluating the large quantity of DNA (GHR 2019a). The assessment of Louis’s DNA
sequences is needed to effectively evaluate their protein-coding regions and their
individualized variations. Whole genome sequencing is the only effective method in Louis’s
case to determine and evaluate his mutations related to oculomotor apraxia and associated
developmental delay/intellectual disability. This genome assessment will substantially assist
in confirming Louis’s oculomotor apraxia and related clinical complications. Whole genome
sequencing technique will also help in evaluating the inheritance pattern of the genes
including APTX, PIK3R5, PNKP, and SETX (Centogene 2019). Resultantly, the technique
will help in tracking the associated disorder like ataxia and determining/ruling-out the risk for
hypoalbuminemia and other similar conditions. Whole genome sequencing the presented case
will assist in ruling out the differential diagnoses including ataxia-telangiectasia, axonal
neuropathy, spinocerebellar ataxia type 2, and coenzyme Q1/vitamin E deficiency
Louis’s oculomotor apraxia along with developmental delay was inherited in an
autosomal recessive format (GHR 2019). Both mutated recessive genes were transferred from
parents to Louis in a manner that his cells contained both mutated genes copy whose overt
expression resulted in the development of oculomotor apraxia. Louis’s parents probably
carried the single version of the mutated recessive gene and that’s why never expressed the
clinical manifestations of oculomotor apraxia with developmental delay. Oculomotor apraxia
in the presented context exhibits 25% risk of its expression in an autosomal recessive pattern
(Coutinho 2019). This indicates that Louis and his sister has a 25% chance of acquiring this
congenital anomaly at their birth. Furthermore, they had a 25% chance of neither acquiring
the disease nor exhibiting the carrier status. Louis and his sister had a 50% probability of
becoming the symptomatic carriers. Louis in the presented scenario qualified the 25%
probability of acquiring the disease and eventually expressed the autosomal recessive pattern
of oculomotor apraxia. The homozygous mutation pattern that arrived from the phenotype
correlations in Louis’s case made him the victim of oculomotor apraxia. Furthermore, the AT
(ataxia) genes based on their compound heterozygosity in the could have promoted the
development of Louis’s AOA (ataxia with oculomotor apraxia) at birth (McKusick & Hartz
2017).
Why Genome Sequencing Might be Appropriate in the Present Situation? – A Critical
Analysis
DNA sequencing assists in tracking the nucleotide sequences in the genetic codes of
individuals. This technique is highly advantageous in terms of evaluating the mechanism of
genetic disorders. Whole genome sequencing assists in determining the genetic variants by
rapidly evaluating the large quantity of DNA (GHR 2019a). The assessment of Louis’s DNA
sequences is needed to effectively evaluate their protein-coding regions and their
individualized variations. Whole genome sequencing is the only effective method in Louis’s
case to determine and evaluate his mutations related to oculomotor apraxia and associated
developmental delay/intellectual disability. This genome assessment will substantially assist
in confirming Louis’s oculomotor apraxia and related clinical complications. Whole genome
sequencing technique will also help in evaluating the inheritance pattern of the genes
including APTX, PIK3R5, PNKP, and SETX (Centogene 2019). Resultantly, the technique
will help in tracking the associated disorder like ataxia and determining/ruling-out the risk for
hypoalbuminemia and other similar conditions. Whole genome sequencing the presented case
will assist in ruling out the differential diagnoses including ataxia-telangiectasia, axonal
neuropathy, spinocerebellar ataxia type 2, and coenzyme Q1/vitamin E deficiency
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

Page 7 of 17
(Centogene 2019). Whole genome sequencing in Louis’s case will prove to be an appropriate
intervention for evaluating the pathogenic allelic variations in the defected genes. For
example, the assessment of PNKP gene’s pathogenic variations will assist in evaluating
polynucleotide kinase 3'-phosphatase’s encoding pattern and the resultant disruption in DNA-
disruption repair function (Scholz et al. 2018). Whole genome sequencing in the presented
scenario will prove to be highly advantageous in terms of assessing the genetic/germline
variants, frameshift mutations, defects in C-terminal amino acids and their relationship with
Louis’s disease pattern. Louis’s case substantiates the Frame Search intervention to
effectively track and explore the protein query sequence and evaluate the genetic variants
through whole-genome sequencing. The use of GSDB (Genome Sequence Database) in the
presented context will assist in exploring the biological information along with the best
possible nucleotide sequences alignments matching with the mutated phenotypes (Harger et
al. 2000). Although the whole genome variants might not thoroughly reveal the biological
pathways of oculomotor apraxia/developmental delay but will provide significant information
regarding Louis’s allele frequency, molecular biological consequence, and their relationship
with the defected phenotypes.
Part – B
Assessment of the Reported Three Variants
The reported variants pertain to the following configuration.
Variant-1, Location (GRCh37): chr12:88483128C>T Location (GRCh38):
chr12:88089351C>T Gene: CEP290 Variant: homozygous missense variant
Variant-2, Location (GRCh37): chr5:37167148C>T Location (GRCh38):
chr5:37167046C>T Gene: C5ORF42 Variant: homozygous splice donor variant
Variant-3, Location (GRCh37): chr13:32945142_32945143delAG Location
(GRCh38): chr13:32371005_32371006delAG Gene: BRCA2 Variant: heterozygous 2
base pair deletion
Molecular Biological Consequence
The autosomal recessive gene 'CEP290' in the variant – 1 provides a range of
instructions is tracked across the 21.32 position of chromosome 12 (long arm) between the
base pair range of 88,049,013-88,142,216 over the cytogenetic region pertaining to 12q21.32
(GHR 2019b). It's defined in terms of Homo sapiens Annotation Release GRCh38.p12 and
109. The gene CEP290 is also known by the names of 3H11Ag, BBS14, CE290_HUMAN,
centrosomal protein 290kDa, centrosomal protein of 290kDa, CT87, CTCL tumor antigen
se2-2, FLJ13615, FLJ21979, JBTS5, JBTS6, KIAA0373, LCA10, MKS4, monoclonal
(Centogene 2019). Whole genome sequencing in Louis’s case will prove to be an appropriate
intervention for evaluating the pathogenic allelic variations in the defected genes. For
example, the assessment of PNKP gene’s pathogenic variations will assist in evaluating
polynucleotide kinase 3'-phosphatase’s encoding pattern and the resultant disruption in DNA-
disruption repair function (Scholz et al. 2018). Whole genome sequencing in the presented
scenario will prove to be highly advantageous in terms of assessing the genetic/germline
variants, frameshift mutations, defects in C-terminal amino acids and their relationship with
Louis’s disease pattern. Louis’s case substantiates the Frame Search intervention to
effectively track and explore the protein query sequence and evaluate the genetic variants
through whole-genome sequencing. The use of GSDB (Genome Sequence Database) in the
presented context will assist in exploring the biological information along with the best
possible nucleotide sequences alignments matching with the mutated phenotypes (Harger et
al. 2000). Although the whole genome variants might not thoroughly reveal the biological
pathways of oculomotor apraxia/developmental delay but will provide significant information
regarding Louis’s allele frequency, molecular biological consequence, and their relationship
with the defected phenotypes.
Part – B
Assessment of the Reported Three Variants
The reported variants pertain to the following configuration.
Variant-1, Location (GRCh37): chr12:88483128C>T Location (GRCh38):
chr12:88089351C>T Gene: CEP290 Variant: homozygous missense variant
Variant-2, Location (GRCh37): chr5:37167148C>T Location (GRCh38):
chr5:37167046C>T Gene: C5ORF42 Variant: homozygous splice donor variant
Variant-3, Location (GRCh37): chr13:32945142_32945143delAG Location
(GRCh38): chr13:32371005_32371006delAG Gene: BRCA2 Variant: heterozygous 2
base pair deletion
Molecular Biological Consequence
The autosomal recessive gene 'CEP290' in the variant – 1 provides a range of
instructions is tracked across the 21.32 position of chromosome 12 (long arm) between the
base pair range of 88,049,013-88,142,216 over the cytogenetic region pertaining to 12q21.32
(GHR 2019b). It's defined in terms of Homo sapiens Annotation Release GRCh38.p12 and
109. The gene CEP290 is also known by the names of 3H11Ag, BBS14, CE290_HUMAN,
centrosomal protein 290kDa, centrosomal protein of 290kDa, CT87, CTCL tumor antigen
se2-2, FLJ13615, FLJ21979, JBTS5, JBTS6, KIAA0373, LCA10, MKS4, monoclonal
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

Page 8 of 17
antibody 3H11 antigen, nephrocystin-6, NPHP6, POC3, POC3 centriolar protein homolog,
prostate cancer antigen T21, rd16, SLSN6, and tumor antigen se2-2 (Wang et al. 2018).
CEP290 substantially contributes to the development of defects including renal
abnormalities, retinal dystrophy, and ocular defects. CEP290 contains the centrosomal
protein and its mutations contribute to the development of Leber’s Congenital Amaurosis
(LCA) with clinical manifestations including roving eye movements, nystagmus, and
pigmentary retinopathy. CEP290 mutations in many scenarios also contribute to the
development of Joubert Syndrome.
C5ORF42 gene in the variant – 2 occupies the 13.2 location of the chromosome-5
(short arm) at the 5p13.2 cytogenetic region (GHR 2019c). The molecular-region of the gene
contains the chromosome-5 with the base pair range pertaining to 37,063,928-37,249,499.
C5ORF42 gene encodes the transmembrane protein with presumably coiled demarcations.
The mutation in C5ORF42 gene induces the development of JBTS (Joubert syndrome).
C5ORF42 gene also induces ciliogenesis and directional cell migration through the induction
of cell polarity. Planar polarity effectors and ciliogenesis (i.e. CPLANE) coordinate with
C5ORF42 gene and influence its functionality. C5ORF42 gene promotes the interaction
between basal bodies and peripheral IFT-A proteins.
The tumor suppression in Homo sapiens BRCA2 gene is caused by the protein
encoded by the BRCA2 gene (GHR 2019d). This gene also repairs the deteriorated DNA
through protein-protein interactions. BRCA2 gene consistently removes the gaps across the
DNA that continue to occur under the impact of chromosomal exchange process or
environmental exposure. BRCA2 gene-based DNA repair mechanism effectively maintains
the stability and consistency of the significant genetic data. The BRCA2 gene is also regarded
as a potential modulator of cytokinesis. BRCA2 mutations significantly increase the risk of
ovarian/breast cancers and epithelial malignancies (Casaubon & Regan 2018). The autosomal
dominant inheritance pattern of BRCA2 mutations substantially elevates the genetic risk of
cancer inheritance. BRCA2 gene is tracked at 13.1 location across the chromosome 13 (long
arm). The BRCA2 gene is molecularly located over chromosome 13 in terms of the base pair
range pertaining to 32,315,480-32,399,672 (GHR 2019d).
Allele Frequency
The hypomorphic-alleles of CEP290 substantially alter the pathophysiology of
heterogeneous retinal ciliopathies. For example, Cep290rd16/+ is a heterozygous
hypomorphic allele that potentially induces retinal dysfunction and degeneration in the
affected beings (Rao et al. 2016). The genetic interaction between the hypomorphic alleles of
antibody 3H11 antigen, nephrocystin-6, NPHP6, POC3, POC3 centriolar protein homolog,
prostate cancer antigen T21, rd16, SLSN6, and tumor antigen se2-2 (Wang et al. 2018).
CEP290 substantially contributes to the development of defects including renal
abnormalities, retinal dystrophy, and ocular defects. CEP290 contains the centrosomal
protein and its mutations contribute to the development of Leber’s Congenital Amaurosis
(LCA) with clinical manifestations including roving eye movements, nystagmus, and
pigmentary retinopathy. CEP290 mutations in many scenarios also contribute to the
development of Joubert Syndrome.
C5ORF42 gene in the variant – 2 occupies the 13.2 location of the chromosome-5
(short arm) at the 5p13.2 cytogenetic region (GHR 2019c). The molecular-region of the gene
contains the chromosome-5 with the base pair range pertaining to 37,063,928-37,249,499.
C5ORF42 gene encodes the transmembrane protein with presumably coiled demarcations.
The mutation in C5ORF42 gene induces the development of JBTS (Joubert syndrome).
C5ORF42 gene also induces ciliogenesis and directional cell migration through the induction
of cell polarity. Planar polarity effectors and ciliogenesis (i.e. CPLANE) coordinate with
C5ORF42 gene and influence its functionality. C5ORF42 gene promotes the interaction
between basal bodies and peripheral IFT-A proteins.
The tumor suppression in Homo sapiens BRCA2 gene is caused by the protein
encoded by the BRCA2 gene (GHR 2019d). This gene also repairs the deteriorated DNA
through protein-protein interactions. BRCA2 gene consistently removes the gaps across the
DNA that continue to occur under the impact of chromosomal exchange process or
environmental exposure. BRCA2 gene-based DNA repair mechanism effectively maintains
the stability and consistency of the significant genetic data. The BRCA2 gene is also regarded
as a potential modulator of cytokinesis. BRCA2 mutations significantly increase the risk of
ovarian/breast cancers and epithelial malignancies (Casaubon & Regan 2018). The autosomal
dominant inheritance pattern of BRCA2 mutations substantially elevates the genetic risk of
cancer inheritance. BRCA2 gene is tracked at 13.1 location across the chromosome 13 (long
arm). The BRCA2 gene is molecularly located over chromosome 13 in terms of the base pair
range pertaining to 32,315,480-32,399,672 (GHR 2019d).
Allele Frequency
The hypomorphic-alleles of CEP290 substantially alter the pathophysiology of
heterogeneous retinal ciliopathies. For example, Cep290rd16/+ is a heterozygous
hypomorphic allele that potentially induces retinal dysfunction and degeneration in the
affected beings (Rao et al. 2016). The genetic interaction between the hypomorphic alleles of

Page 9 of 17
CEP290 and RPGR leads to the establishment of heterogeneous retinal ciliopathies. The
predomination of CEP290 (AHI1) modifier alleles induces LCA and associated clinical
manifestations (Sfari_Gene 2019). The in-frame deletion and frameshift/non-sense
deleterious mutations contribute to the development of early-onset retinal degeneration and
retinitis pigmentosa/congenital amaurosis (den-Hollander et al. 2006). The molecular
assessment in a recently reported study revealed the attribution of CEP-290 allele ‘c.6012-
12T>A (75.0%)’ to the development of Joubert Syndrome (Suzuki et al. 2016). Several
alleles of the CEP290 gene undergo c.2991+1655A→G mutation that attributes to
approximately 21% of Leber’s Congenital Amaurosis scenarios (den-Hollander et al. 2006).
C5ORF42 associates with CPLANE1 to configure 11 allelic variants (OMIM 2019).
Allelic variants including ARG1336TRP, IVS35DS, G-A, +1, 1-BP DEL, NT6407,
ARG1602TER, and ARG2493TER correspond to Joubert Syndrome 17. The significance of
the variant ALA1564THR is unknown. However, the variants including IVS18AS, G-T, -1,
1-BP DEL, 493A, IVS19AS, A-G, -2, SER1127LEU, and ASP1287HIS correspond to
orofacial syndrome VI. The high frequency of mutations in C5ORF42 alleles leads to the
expression of JBS and its clinical manifestations (including intellectual disability and short
stature). These significant mutations include c.4690G>A [p.Ala1564Thr]), c.7400+1G>A,
and c.4006C>T [p.Arg1336Trp] (Srour et al. 2012). The appearance of these mutations leads
to the induction of neurodevelopmental disorder in the affected individuals. These findings
indicate that the expression of any of these mutated alleles in Louis’s case could have
resulted in the development of his neurodevelopmental manifestations.
The BRCA2 alleles exhibit copy number reduction in ratios of 4:3, 3:2, and 4:2 across
the selected loci that affirms the physical deletion of the wild type alleles’ heterozygosity
(Staff et al. 2000). This heterozygosity loss is followed by polyploidization that resultantly
leads to the replication of the mutant allele. The wild type allelic loss reciprocates with the
processes like gene conversion, non-disjunctional chromosomal elimination, and mitotic
recombination. Genetically predisposed people likely develop BRCA2-based tumors while
incorporating additional copy numbers of the allele mutant.
Likely Pathogenicity
The protein encoded by CEP290 gene (i.e. centrosomal protein 290) contains thirteen
coiled regions homologous with motif A (ATP/GTP binding location), tropomyosin
homology regions, 6-KID motifs, and ATPases (with SMC chromosome segregation)
(Gene_ID_80184_NCBI 2018). The protein encoded by CEP290 is confined to cilia and
centrosome with locations embedded for amidation, N-myristoylation, phosphorylation,
CEP290 and RPGR leads to the establishment of heterogeneous retinal ciliopathies. The
predomination of CEP290 (AHI1) modifier alleles induces LCA and associated clinical
manifestations (Sfari_Gene 2019). The in-frame deletion and frameshift/non-sense
deleterious mutations contribute to the development of early-onset retinal degeneration and
retinitis pigmentosa/congenital amaurosis (den-Hollander et al. 2006). The molecular
assessment in a recently reported study revealed the attribution of CEP-290 allele ‘c.6012-
12T>A (75.0%)’ to the development of Joubert Syndrome (Suzuki et al. 2016). Several
alleles of the CEP290 gene undergo c.2991+1655A→G mutation that attributes to
approximately 21% of Leber’s Congenital Amaurosis scenarios (den-Hollander et al. 2006).
C5ORF42 associates with CPLANE1 to configure 11 allelic variants (OMIM 2019).
Allelic variants including ARG1336TRP, IVS35DS, G-A, +1, 1-BP DEL, NT6407,
ARG1602TER, and ARG2493TER correspond to Joubert Syndrome 17. The significance of
the variant ALA1564THR is unknown. However, the variants including IVS18AS, G-T, -1,
1-BP DEL, 493A, IVS19AS, A-G, -2, SER1127LEU, and ASP1287HIS correspond to
orofacial syndrome VI. The high frequency of mutations in C5ORF42 alleles leads to the
expression of JBS and its clinical manifestations (including intellectual disability and short
stature). These significant mutations include c.4690G>A [p.Ala1564Thr]), c.7400+1G>A,
and c.4006C>T [p.Arg1336Trp] (Srour et al. 2012). The appearance of these mutations leads
to the induction of neurodevelopmental disorder in the affected individuals. These findings
indicate that the expression of any of these mutated alleles in Louis’s case could have
resulted in the development of his neurodevelopmental manifestations.
The BRCA2 alleles exhibit copy number reduction in ratios of 4:3, 3:2, and 4:2 across
the selected loci that affirms the physical deletion of the wild type alleles’ heterozygosity
(Staff et al. 2000). This heterozygosity loss is followed by polyploidization that resultantly
leads to the replication of the mutant allele. The wild type allelic loss reciprocates with the
processes like gene conversion, non-disjunctional chromosomal elimination, and mitotic
recombination. Genetically predisposed people likely develop BRCA2-based tumors while
incorporating additional copy numbers of the allele mutant.
Likely Pathogenicity
The protein encoded by CEP290 gene (i.e. centrosomal protein 290) contains thirteen
coiled regions homologous with motif A (ATP/GTP binding location), tropomyosin
homology regions, 6-KID motifs, and ATPases (with SMC chromosome segregation)
(Gene_ID_80184_NCBI 2018). The protein encoded by CEP290 is confined to cilia and
centrosome with locations embedded for amidation, N-myristoylation, phosphorylation,
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

Page 10 of 17
tyrosine sulfation, and N-glycosylation. Potential mutations in the CEP290 gene lead to the
development of nephronophthisis and Joubert syndrome. The antibodies’ pattern induced by
CEP290 protein is indicative of numerous types of carcinomatous conditions.
C5ORF42 transcript’s expression in the human testis and brain contributes to the
pathogenicity of this gene (OMIM 2019). The developmental defects related to ciliopathy
emanate reportedly occur under the impact of morpholino oligonucleotides-based knockdown
of C5ORF42 gene. These defects include the defective left-right pattern, disrupted Hedgehog
pattern, and neural tube closure failure (OMIM 2019). The mutations in the exome
sequencing of C5ORF42 gene lead to the induction of global developmental delay, mild
intellectual disability or borderline intelligence in the affected children. These mutations also
contribute to syndactyly, postaxial polydactyly, limb abnormalities, episodic
hyperventilation, breathing abnormalities, and oculomotor apraxia. However, the two-slice,
five missense, three nonsense, and four frameshift mutations in the C5ORF42 gene lead to
the development/expression of OFD6 (Orofaciodigital Syndrome VI) in the affected
individuals (OMIM 2019).
350 SNPs-based carriers of BRCA2 alleles exhibit elevated risk of breast cancer and
its clinical manifestations (OMIM 2019a). The BRCA2 carriers who exhibit FBXL7
(rs12652447) and LOC134997 (rs9393597) experience a severe predisposition towards breast
cancer. BRCA2 deleterious germline mutations in males pertaining to the age of 56 years or
above elevate the 23-fold risk of prostate cancer (OMIM 2019a). A homozygous elimination
at the BRCA2 locus containing 6-cM location and chromosome 13q12.3 (1-cM region) leads
to the development of human pancreatic adenocarcinoma. Similarly, elimination of BRCA2-
based Mb 13q12.3 locus leads to the development of CLL (chronic lymphocytic leukemia)
(OMIM 2019a). Balletic BRCA2 mutations are categorized in accordance with their
functional impact and capacity to cause breast malignancies in predisposed heterozygous
individuals.
Diagnoses and their Relevance to Louis' Phenotypes
CEP290 gene mutations contribute to the diagnoses including Leber congenital
amaurosis, Bardet-Biedl syndrome, Joubert syndrome, Meckel syndrome, Senior-Løken
syndrome (i.e. ciliopathies) (GHR 2019b). Most of these disease conditions potentially
impact the renal system, retina, and central nervous system (i.e. spinal cord and brain). The
abnormal CEP290 protein version presumably impacts the functionality of cilia that leads to
the development of clinical manifestations. Bardet-Biedl syndrome’s manifestations include
polydactyly and vision loss. The ocular manifestations of this disease are somewhat related to
tyrosine sulfation, and N-glycosylation. Potential mutations in the CEP290 gene lead to the
development of nephronophthisis and Joubert syndrome. The antibodies’ pattern induced by
CEP290 protein is indicative of numerous types of carcinomatous conditions.
C5ORF42 transcript’s expression in the human testis and brain contributes to the
pathogenicity of this gene (OMIM 2019). The developmental defects related to ciliopathy
emanate reportedly occur under the impact of morpholino oligonucleotides-based knockdown
of C5ORF42 gene. These defects include the defective left-right pattern, disrupted Hedgehog
pattern, and neural tube closure failure (OMIM 2019). The mutations in the exome
sequencing of C5ORF42 gene lead to the induction of global developmental delay, mild
intellectual disability or borderline intelligence in the affected children. These mutations also
contribute to syndactyly, postaxial polydactyly, limb abnormalities, episodic
hyperventilation, breathing abnormalities, and oculomotor apraxia. However, the two-slice,
five missense, three nonsense, and four frameshift mutations in the C5ORF42 gene lead to
the development/expression of OFD6 (Orofaciodigital Syndrome VI) in the affected
individuals (OMIM 2019).
350 SNPs-based carriers of BRCA2 alleles exhibit elevated risk of breast cancer and
its clinical manifestations (OMIM 2019a). The BRCA2 carriers who exhibit FBXL7
(rs12652447) and LOC134997 (rs9393597) experience a severe predisposition towards breast
cancer. BRCA2 deleterious germline mutations in males pertaining to the age of 56 years or
above elevate the 23-fold risk of prostate cancer (OMIM 2019a). A homozygous elimination
at the BRCA2 locus containing 6-cM location and chromosome 13q12.3 (1-cM region) leads
to the development of human pancreatic adenocarcinoma. Similarly, elimination of BRCA2-
based Mb 13q12.3 locus leads to the development of CLL (chronic lymphocytic leukemia)
(OMIM 2019a). Balletic BRCA2 mutations are categorized in accordance with their
functional impact and capacity to cause breast malignancies in predisposed heterozygous
individuals.
Diagnoses and their Relevance to Louis' Phenotypes
CEP290 gene mutations contribute to the diagnoses including Leber congenital
amaurosis, Bardet-Biedl syndrome, Joubert syndrome, Meckel syndrome, Senior-Løken
syndrome (i.e. ciliopathies) (GHR 2019b). Most of these disease conditions potentially
impact the renal system, retina, and central nervous system (i.e. spinal cord and brain). The
abnormal CEP290 protein version presumably impacts the functionality of cilia that leads to
the development of clinical manifestations. Bardet-Biedl syndrome’s manifestations include
polydactyly and vision loss. The ocular manifestations of this disease are somewhat related to
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

Page 11 of 17
uncontrolled Louis’s eye movements (GHR 2019b). Resultantly, Louis also experiences the
risk of vision loss at a later point in time. Similarly, Joubert syndrome exhibits the
characteristics of oculomotor apraxia and ataxia along with the intellectual disability and
delayed development (as experienced by Louis) (GHR 2019b). Meckel syndrome matches
with Louis phenotypes in a manner that it impacts the brain/eye development processes.
Mutations in C5ORF42 gene lead to the development of OFD6 (Orofaciodigital
syndrome 6) and JBTS17 (Joubert syndrome 17) (GHR 2019c). OFD6 leads to the subtypes’
delineation along with malformations of digits, face, and oral cavity. The phenotypic
characteristics of OFD6 that match with Louis’s phenotypes include the brain/central nervous
system manifestations. However, the other phenotypic abnormalities include central
polydactyly, metacarpal abnormalities, and defects related to upper lip notch, frenula, tongue
hamartomas, and molar tooth sign (GHR 2019c). Furthermore, the psychomotor delay,
oculomotor apraxia, and cerebellar ataxia exhibited by JBTS17 radically match with Louis’s
reported phenotypes. Other phenotypes exhibited by JBTS17 include the renal disease and
retinal dystrophy (GHR 2019c).
BRCA2 gene mutations mostly lead to the development of breast cancer, ovarian
cancer, prostate cancer, Fanconi anemia, and melanoma (GHR 2019d). The non-functional
BRCA2 protein version leads to the development of breast cancer and its phenotypic
characteristics including skin irritation, retraction/tenderness or nipple discharge. Louis does
not possess any of these phenotypic features; however, experiences risk of cancer-based on
his family history (of pancreatic cancer). BRCA2 gene mutations lead to the development of
prostate cancer complications including pain with ejaculation, hematuria, and urinary
dribbling. Louis experiences risk of developing these phenotypes based on his risk of
undergoing BRCA2 mutations (GHR 2019d). BRCA2 gene mutations elevate the risk of
Fanconi anemia and associated clinical complications of forearm/thumb malformation,
hypopigmentation, cardiac defect, hearing loss, and gastrointestinal abnormality. Fanconi
anemia also induces the phenotypic defects related to spinal cord and brain (i.e. central
nervous system [CNS]). Louis’s presently reported phenotypic abnormalities somewhat
match with the CNS manifestations (including developmental delay) (GHR 2019d).
Ethical Implications for Family Members
The greatest ethical implications of the genetic testing in Louis’s case relate to the fact
that the whole genome sequencing might not retrieve definitive outcomes (NSW 2018). The
acquisition of informed consent is highly necessary for undertaking the genetic analysis.
However, Louis in the presented scenario might not be able to consciously provide informed
uncontrolled Louis’s eye movements (GHR 2019b). Resultantly, Louis also experiences the
risk of vision loss at a later point in time. Similarly, Joubert syndrome exhibits the
characteristics of oculomotor apraxia and ataxia along with the intellectual disability and
delayed development (as experienced by Louis) (GHR 2019b). Meckel syndrome matches
with Louis phenotypes in a manner that it impacts the brain/eye development processes.
Mutations in C5ORF42 gene lead to the development of OFD6 (Orofaciodigital
syndrome 6) and JBTS17 (Joubert syndrome 17) (GHR 2019c). OFD6 leads to the subtypes’
delineation along with malformations of digits, face, and oral cavity. The phenotypic
characteristics of OFD6 that match with Louis’s phenotypes include the brain/central nervous
system manifestations. However, the other phenotypic abnormalities include central
polydactyly, metacarpal abnormalities, and defects related to upper lip notch, frenula, tongue
hamartomas, and molar tooth sign (GHR 2019c). Furthermore, the psychomotor delay,
oculomotor apraxia, and cerebellar ataxia exhibited by JBTS17 radically match with Louis’s
reported phenotypes. Other phenotypes exhibited by JBTS17 include the renal disease and
retinal dystrophy (GHR 2019c).
BRCA2 gene mutations mostly lead to the development of breast cancer, ovarian
cancer, prostate cancer, Fanconi anemia, and melanoma (GHR 2019d). The non-functional
BRCA2 protein version leads to the development of breast cancer and its phenotypic
characteristics including skin irritation, retraction/tenderness or nipple discharge. Louis does
not possess any of these phenotypic features; however, experiences risk of cancer-based on
his family history (of pancreatic cancer). BRCA2 gene mutations lead to the development of
prostate cancer complications including pain with ejaculation, hematuria, and urinary
dribbling. Louis experiences risk of developing these phenotypes based on his risk of
undergoing BRCA2 mutations (GHR 2019d). BRCA2 gene mutations elevate the risk of
Fanconi anemia and associated clinical complications of forearm/thumb malformation,
hypopigmentation, cardiac defect, hearing loss, and gastrointestinal abnormality. Fanconi
anemia also induces the phenotypic defects related to spinal cord and brain (i.e. central
nervous system [CNS]). Louis’s presently reported phenotypic abnormalities somewhat
match with the CNS manifestations (including developmental delay) (GHR 2019d).
Ethical Implications for Family Members
The greatest ethical implications of the genetic testing in Louis’s case relate to the fact
that the whole genome sequencing might not retrieve definitive outcomes (NSW 2018). The
acquisition of informed consent is highly necessary for undertaking the genetic analysis.
However, Louis in the presented scenario might not be able to consciously provide informed

Page 12 of 17
consent while understanding the ethical implications. Furthermore, genomic analysis has the
risk of unwanted coercion or persuasion by the patient’s family members or the researcher for
undergoing the recommended assessment (NSW 2018). The unauthorized sharing of genetic
findings could pose serious confidentiality breach issues and discredit the genomic testing or
treatment approaches.
Further Testing or Treatment Implications for Louis and Other Family Members
Louis’s nieces need to undergo OAAASE (Oculomotor Apraxia Ataxia Advanced
Sequencing Evaluation) with the objective of predictively or pre-symptomatically evaluating
EAOH (Ataxia-oculomotor apraxia type 1), ALS4 (Amyotrophic lateral sclerosis type 4), and
SCAR1 (Spinocerebellar ataxia autosomal recessive 1) (NCBI_GTR 2018). The assessment
of APTX (9p21.1) gene will be required to evaluate EAOH. However, SETX (9q34.13) gene
evaluation will help in determining/ruling out ALS4 and SCAR1. The coding regions’
sequence assessment and MPS (massively parallel sequencing) will help in undertaking the
concerned genetic assessments. Louis and his family members will also require undertaking
the genetic testing/whole genome sequencing of STK11, ATM, PALB2, CDKN2A, PALB2,
and BRCA1 genes because of their reported familial pancreatic cancer and an associated risk
of mutations related to PRSS1/germline cationic trypsinogen (ASCO 2018) (Rustgi 2014).
consent while understanding the ethical implications. Furthermore, genomic analysis has the
risk of unwanted coercion or persuasion by the patient’s family members or the researcher for
undergoing the recommended assessment (NSW 2018). The unauthorized sharing of genetic
findings could pose serious confidentiality breach issues and discredit the genomic testing or
treatment approaches.
Further Testing or Treatment Implications for Louis and Other Family Members
Louis’s nieces need to undergo OAAASE (Oculomotor Apraxia Ataxia Advanced
Sequencing Evaluation) with the objective of predictively or pre-symptomatically evaluating
EAOH (Ataxia-oculomotor apraxia type 1), ALS4 (Amyotrophic lateral sclerosis type 4), and
SCAR1 (Spinocerebellar ataxia autosomal recessive 1) (NCBI_GTR 2018). The assessment
of APTX (9p21.1) gene will be required to evaluate EAOH. However, SETX (9q34.13) gene
evaluation will help in determining/ruling out ALS4 and SCAR1. The coding regions’
sequence assessment and MPS (massively parallel sequencing) will help in undertaking the
concerned genetic assessments. Louis and his family members will also require undertaking
the genetic testing/whole genome sequencing of STK11, ATM, PALB2, CDKN2A, PALB2,
and BRCA1 genes because of their reported familial pancreatic cancer and an associated risk
of mutations related to PRSS1/germline cationic trypsinogen (ASCO 2018) (Rustgi 2014).
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide
1 out of 17
Your All-in-One AI-Powered Toolkit for Academic Success.
+13062052269
info@desklib.com
Available 24*7 on WhatsApp / Email
![[object Object]](/_next/static/media/star-bottom.7253800d.svg)
Unlock your academic potential
Copyright © 2020–2025 A2Z Services. All Rights Reserved. Developed and managed by ZUCOL.