Introduction to Regulatory Affairs: US Biological Product Registration
VerifiedAdded on 2023/04/10
|10
|2025
|373
Project
AI Summary
This project provides a comprehensive overview of the US regulatory process for registering innovative biological products. It begins with a classification of new drugs and delves into the functions and activities of the Food and Drug Administration (FDA), the primary regulatory body. The document then outlines the New Drug Application (NDA) process, detailing the steps involved, including pre-clinical and clinical trials (Phases 1, 2, and 3), and the importance of current good manufacturing practices (cGMP). It also specifies the typical time frame for drug approval. The project further examines the regulatory pathway for generic medicinal products, specifically the Abbreviated New Drug Application (ANDA). The project includes references to support the claims and provide further reading on the topic.

Registration of an innovative biological product in US
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

Contents
Classification of the new drug.........................................................................................................3
Functions and activities of FDA......................................................................................................3
Registration process.........................................................................................................................3
Clinical trials....................................................................................................................................5
Phase 1.........................................................................................................................................5
Phase 2.........................................................................................................................................6
Phase 3.........................................................................................................................................6
Current good manufacturing practices (cGMP)..............................................................................6
Time-period.....................................................................................................................................7
For generic medical product............................................................................................................7
Reference.........................................................................................................................................8
Classification of the new drug.........................................................................................................3
Functions and activities of FDA......................................................................................................3
Registration process.........................................................................................................................3
Clinical trials....................................................................................................................................5
Phase 1.........................................................................................................................................5
Phase 2.........................................................................................................................................6
Phase 3.........................................................................................................................................6
Current good manufacturing practices (cGMP)..............................................................................6
Time-period.....................................................................................................................................7
For generic medical product............................................................................................................7
Reference.........................................................................................................................................8

Classification of the new drug
The application for an innovative biological product comes under New Drug Application. There
are two types of NDA and these are 505(b)(1) and 505(b)(2) (FDA, 2019b). The 505(b)(1) is
used for approval of a new product whose active ingredients have not been approved previously.
505(b)(2) pathway on the other hand is used for products that contain similar active ingredients
as a previously approved product (The Weinberg Group, 2019). The following pages examine
the process of registering an innovative biological product in the United States considering the
505(b)(1) submission type under NDA in form Form FDA-356h (FDA. 2019a).
Functions and activities of FDA
The FDA or the Food and Drug Administration have the responsibility of protecting public
health. FDA thus ensures that medical devices, drugs, biological products, cosmetics, radiation
emitting products and food supply are secured, safe and efficient (FDA, 2019). It also has the
responsibility to advance public health through speeding up of innovations that increases the
effectiveness, safety and affordability of medical products. FDA ensures that the public have
access to accurate, science based information about the foods and drugs so that they can maintain
and improve their heath.
Registration process
The development of a new drug starts with discovery. After new components have been
discovered prototypes are made. Before the newly developed drug can be tested on humans or in
other words can be passed through clinical trials, it has to shown to be safe and effective. This is
done through pre-clinical trials which are conducted on animals (Woodcock and Woosley,
2008). The IRB or the Institutional Review Board present in private clinical research centers,
hospitals, etc. gives the approval for both the pre-clinical and clinical trials. During these pre-
clinical trials the boundaries for the safe use of the treatment is established before the drug can
be tested on humans during clinical trials. After the pre-clinical trials are conducted on animals
in accordance with the Good Laboratory Practices (GLPs), the results of the same are
documented in technical reports or scientific publications (Aziz, 2017). These documents from
part of a report that has to be submitted before approval for commencing with clinical trials can
be availed.
The application for an innovative biological product comes under New Drug Application. There
are two types of NDA and these are 505(b)(1) and 505(b)(2) (FDA, 2019b). The 505(b)(1) is
used for approval of a new product whose active ingredients have not been approved previously.
505(b)(2) pathway on the other hand is used for products that contain similar active ingredients
as a previously approved product (The Weinberg Group, 2019). The following pages examine
the process of registering an innovative biological product in the United States considering the
505(b)(1) submission type under NDA in form Form FDA-356h (FDA. 2019a).
Functions and activities of FDA
The FDA or the Food and Drug Administration have the responsibility of protecting public
health. FDA thus ensures that medical devices, drugs, biological products, cosmetics, radiation
emitting products and food supply are secured, safe and efficient (FDA, 2019). It also has the
responsibility to advance public health through speeding up of innovations that increases the
effectiveness, safety and affordability of medical products. FDA ensures that the public have
access to accurate, science based information about the foods and drugs so that they can maintain
and improve their heath.
Registration process
The development of a new drug starts with discovery. After new components have been
discovered prototypes are made. Before the newly developed drug can be tested on humans or in
other words can be passed through clinical trials, it has to shown to be safe and effective. This is
done through pre-clinical trials which are conducted on animals (Woodcock and Woosley,
2008). The IRB or the Institutional Review Board present in private clinical research centers,
hospitals, etc. gives the approval for both the pre-clinical and clinical trials. During these pre-
clinical trials the boundaries for the safe use of the treatment is established before the drug can
be tested on humans during clinical trials. After the pre-clinical trials are conducted on animals
in accordance with the Good Laboratory Practices (GLPs), the results of the same are
documented in technical reports or scientific publications (Aziz, 2017). These documents from
part of a report that has to be submitted before approval for commencing with clinical trials can
be availed.
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide
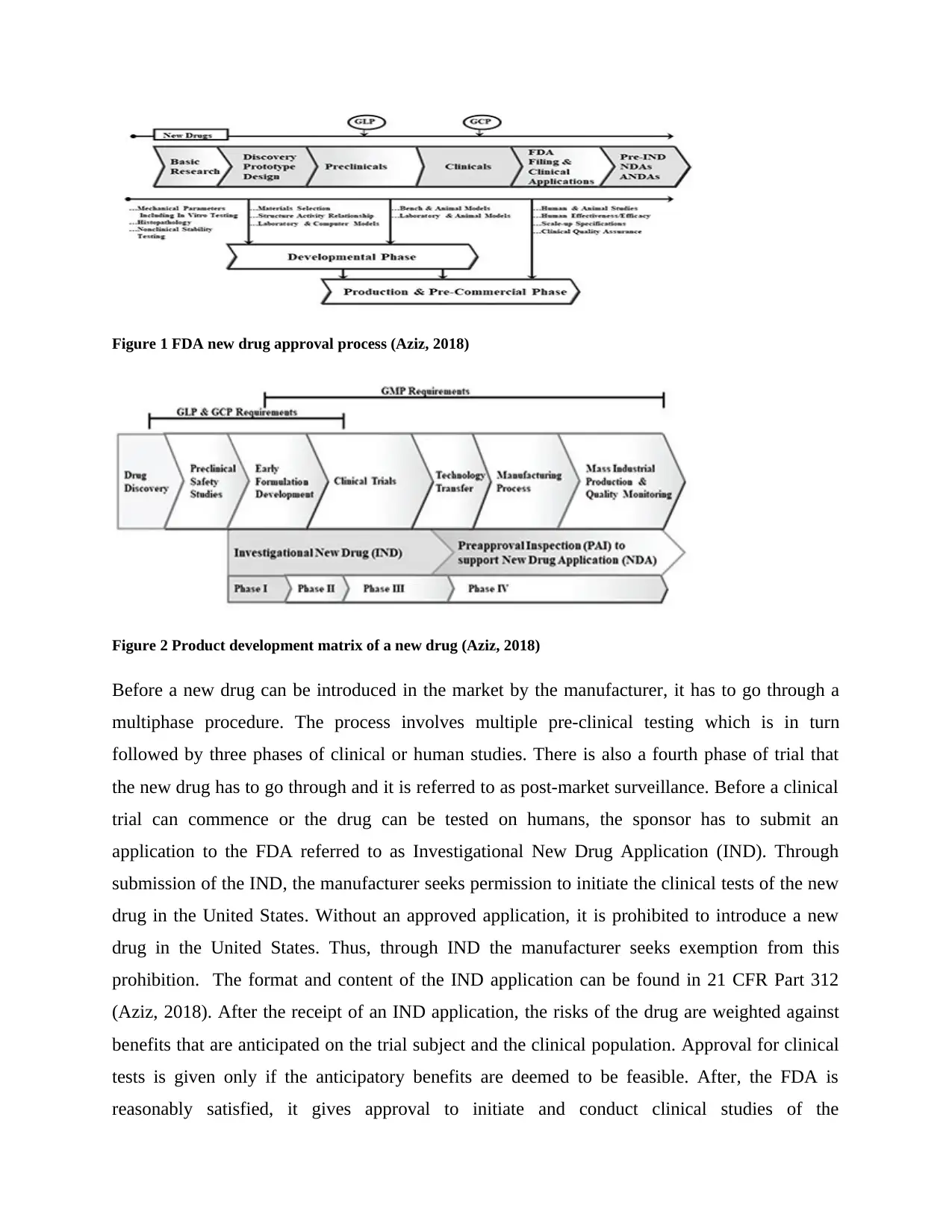
Figure 1 FDA new drug approval process (Aziz, 2018)
Figure 2 Product development matrix of a new drug (Aziz, 2018)
Before a new drug can be introduced in the market by the manufacturer, it has to go through a
multiphase procedure. The process involves multiple pre-clinical testing which is in turn
followed by three phases of clinical or human studies. There is also a fourth phase of trial that
the new drug has to go through and it is referred to as post-market surveillance. Before a clinical
trial can commence or the drug can be tested on humans, the sponsor has to submit an
application to the FDA referred to as Investigational New Drug Application (IND). Through
submission of the IND, the manufacturer seeks permission to initiate the clinical tests of the new
drug in the United States. Without an approved application, it is prohibited to introduce a new
drug in the United States. Thus, through IND the manufacturer seeks exemption from this
prohibition. The format and content of the IND application can be found in 21 CFR Part 312
(Aziz, 2018). After the receipt of an IND application, the risks of the drug are weighted against
benefits that are anticipated on the trial subject and the clinical population. Approval for clinical
tests is given only if the anticipatory benefits are deemed to be feasible. After, the FDA is
reasonably satisfied, it gives approval to initiate and conduct clinical studies of the
Figure 2 Product development matrix of a new drug (Aziz, 2018)
Before a new drug can be introduced in the market by the manufacturer, it has to go through a
multiphase procedure. The process involves multiple pre-clinical testing which is in turn
followed by three phases of clinical or human studies. There is also a fourth phase of trial that
the new drug has to go through and it is referred to as post-market surveillance. Before a clinical
trial can commence or the drug can be tested on humans, the sponsor has to submit an
application to the FDA referred to as Investigational New Drug Application (IND). Through
submission of the IND, the manufacturer seeks permission to initiate the clinical tests of the new
drug in the United States. Without an approved application, it is prohibited to introduce a new
drug in the United States. Thus, through IND the manufacturer seeks exemption from this
prohibition. The format and content of the IND application can be found in 21 CFR Part 312
(Aziz, 2018). After the receipt of an IND application, the risks of the drug are weighted against
benefits that are anticipated on the trial subject and the clinical population. Approval for clinical
tests is given only if the anticipatory benefits are deemed to be feasible. After, the FDA is
reasonably satisfied, it gives approval to initiate and conduct clinical studies of the
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

investigational drug. The clinical study helps to gather evidence of the effectiveness and safety
of the new drug in humans. During these studies, FDA conducts several meetings with the
manufacturer of the new drug (Aziz, 2018).
Clinical trials
In the process of discovery and development of a new drug, clinical trial is very important. FDA
needs to evaluate and review the clinical trial before the new drug can be brought to the market.
Together, the pre-clinical and clinical trials help to demonstrate the safety and effectiveness of
the new drug and are mandatory requirements before filing New Drug Application (NDA) with
the FDA. Careful planning and consideration of the subjects are two important conditions for a
clinical trial (Kaitin, 2010). Clinical trials help in testing the hypothesis and reach conclusion as
to whether the new drug being developed has any effect on the human body and the medical
condition which is being treated. It is also important to consider whether the administration of
the drug can improve the quality of life and health of the subject, has distinct advantage over
current treatment method and can be administered safely. It is important to ensure that the risks
to the participants in the clinical trial are controlled by the sponsor or manufacturer of the drug.
Additionally, rules and regulations that govern protection of the human beings while evaluating
the effectiveness of the new drug should be carefully understood and adhered to during clinical
trials.
Clinical trials are important for the fact that these protocol-designed studies provide data and
information that supports the submission of NDA to the FDA. There are different phases or
stages of the clinical trial depending on the stage in the product life cycle. Normally, clinical
trials are classified into three distinct phases.
Phase 1
The purpose of this phase of the clinical trial is to gather initial information regarding the safety
of the drug and is conducted on a small population of less than 100 individuals. The purpose of
the phase 1 of the clinical study is to determine the safe dosage of the drug that can be
administered so as to be effective and be least toxic to the patient (Kaitin and DiMasi, 2011). In
this phase, tests are conducted to establish relation between plasma concentration level and the
of the new drug in humans. During these studies, FDA conducts several meetings with the
manufacturer of the new drug (Aziz, 2018).
Clinical trials
In the process of discovery and development of a new drug, clinical trial is very important. FDA
needs to evaluate and review the clinical trial before the new drug can be brought to the market.
Together, the pre-clinical and clinical trials help to demonstrate the safety and effectiveness of
the new drug and are mandatory requirements before filing New Drug Application (NDA) with
the FDA. Careful planning and consideration of the subjects are two important conditions for a
clinical trial (Kaitin, 2010). Clinical trials help in testing the hypothesis and reach conclusion as
to whether the new drug being developed has any effect on the human body and the medical
condition which is being treated. It is also important to consider whether the administration of
the drug can improve the quality of life and health of the subject, has distinct advantage over
current treatment method and can be administered safely. It is important to ensure that the risks
to the participants in the clinical trial are controlled by the sponsor or manufacturer of the drug.
Additionally, rules and regulations that govern protection of the human beings while evaluating
the effectiveness of the new drug should be carefully understood and adhered to during clinical
trials.
Clinical trials are important for the fact that these protocol-designed studies provide data and
information that supports the submission of NDA to the FDA. There are different phases or
stages of the clinical trial depending on the stage in the product life cycle. Normally, clinical
trials are classified into three distinct phases.
Phase 1
The purpose of this phase of the clinical trial is to gather initial information regarding the safety
of the drug and is conducted on a small population of less than 100 individuals. The purpose of
the phase 1 of the clinical study is to determine the safe dosage of the drug that can be
administered so as to be effective and be least toxic to the patient (Kaitin and DiMasi, 2011). In
this phase, tests are conducted to establish relation between plasma concentration level and the

dosage of drug to establish the pharmacokinetics and safety of the drug in question. As the
results of the Phase 1 of the clinical trial, Phase 2 of the clinical trial is developed.
Phase 2
Compared to phase 1, the phase 2 is performed on a large number of people, typically between
100 and 300, who are suffering from the disease that the drug is meant to treat. The phase 2 is a
dose ranging study that is used to determine the minimum and maximum effective dose of the
drug being tested (Kneller, 2010). There are two subparts of the Phase 2 referred to as phase 2a
and phase 2b. Phase 2a which is the pilot study helps in determining the initial effectiveness.
Phase 2b is controlled study that is conducted on several hundred people. Before, the evaluation
of the dose regimen in the phase 3 trial, there should be availability of sufficient data in regards
to efficiency and tolerability of different dose requirements as part of the phase 2 trials. Evidence
to establish that the mechanism of action evidenced in animals is also evidenced in humans is
established in this phase. After conclusion of phase 2, the sponsor and FDA come together to
discuss the plan and data for phase 3.
Phase 3
Phase 3 of the clinical trial is considered to be the most important and is designed for collecting
all the necessary data to meet the requirements of safety and effectiveness of a new drug before
the same is approved for application in the US market (Watkins, 2011). These studies are very
large and are conducted on a population of thousands at multiple locations. Such studies are
conducted in random manner. As compared to control treatments, detailed data are gathered
regarding the effectiveness of the new drug compound. In order to evaluate the side effects and
safety of the new drug, subjects are often tracked. Phase 3 is also used to determine effectiveness
of the drug, its stability, conditions for storage, labeling and marketing claims. All the clinical
study are concluded after the completion of phase 3 and the manufacturer can submit the NDA to
the FDA to gain the premarket approval for the new drug in United States.
Current good manufacturing practices (cGMP)
The cGMPs are part of the quality assurance process that ensures that the drug is consistently
controlled and produced in accordance with the quality standards. The United States adopted
ICHQ10 in the year 2009. By implementing the ICHQ10 throughout the PLC, the link between
results of the Phase 1 of the clinical trial, Phase 2 of the clinical trial is developed.
Phase 2
Compared to phase 1, the phase 2 is performed on a large number of people, typically between
100 and 300, who are suffering from the disease that the drug is meant to treat. The phase 2 is a
dose ranging study that is used to determine the minimum and maximum effective dose of the
drug being tested (Kneller, 2010). There are two subparts of the Phase 2 referred to as phase 2a
and phase 2b. Phase 2a which is the pilot study helps in determining the initial effectiveness.
Phase 2b is controlled study that is conducted on several hundred people. Before, the evaluation
of the dose regimen in the phase 3 trial, there should be availability of sufficient data in regards
to efficiency and tolerability of different dose requirements as part of the phase 2 trials. Evidence
to establish that the mechanism of action evidenced in animals is also evidenced in humans is
established in this phase. After conclusion of phase 2, the sponsor and FDA come together to
discuss the plan and data for phase 3.
Phase 3
Phase 3 of the clinical trial is considered to be the most important and is designed for collecting
all the necessary data to meet the requirements of safety and effectiveness of a new drug before
the same is approved for application in the US market (Watkins, 2011). These studies are very
large and are conducted on a population of thousands at multiple locations. Such studies are
conducted in random manner. As compared to control treatments, detailed data are gathered
regarding the effectiveness of the new drug compound. In order to evaluate the side effects and
safety of the new drug, subjects are often tracked. Phase 3 is also used to determine effectiveness
of the drug, its stability, conditions for storage, labeling and marketing claims. All the clinical
study are concluded after the completion of phase 3 and the manufacturer can submit the NDA to
the FDA to gain the premarket approval for the new drug in United States.
Current good manufacturing practices (cGMP)
The cGMPs are part of the quality assurance process that ensures that the drug is consistently
controlled and produced in accordance with the quality standards. The United States adopted
ICHQ10 in the year 2009. By implementing the ICHQ10 throughout the PLC, the link between
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

manufacturing activities and drug development process is strengthened. ISBN 0273 -3099 which
is the Industry sponsored guideline for continuous quality improvement has also been adopted by
FDA (Aziz, 2018). Instead of a detail prescription as to how a drug needs to be designed and
manufactured, the cGMP regulations presents a framework for the manufacturers to develop and
follow. The most important consideration behind GMP regulations is the fact that it is necessary
to design and build quality in the product.
Figure 3 Flow chart for inspection under GMP (Aziz, 2018)
Time-period
As mentioned there are two types of trials that must be conducted before approval of a new drug.
The pre-clinical trial takes around 4 years to complete. On successful completion of pre-clinical
trial IND application is submitted to FDA. Upon approval of IND, the clinical trials commence
with phase 1, 2 and 3 and in total take 1-3 years to complete (OMNICS International, 2019). On
an average it would take around 8 to 12 years for the approval of a new drug in United States.
For generic medical product
Generic medical products are not subject to either pre-clinical or clinical trials to establish their
safety and effectiveness. This is because the safety and effectiveness of the drug compound was
already established by the new drug or the innovative biological product (of which the generic
drug is bioequivalent) through the process of NDA. Instead the manufacturer of a generic drug
has to file Abbreviated New Drug Application (ANDA). ANDA is for all generic products which
are bioequivalent or pharmaceutical equivalent of an existing product that has been approved
through the NDA process. The manufacturer or the sponsor of the generic drug provides the
required data and information through ANDA to establish that the product is bioequivalent of a
is the Industry sponsored guideline for continuous quality improvement has also been adopted by
FDA (Aziz, 2018). Instead of a detail prescription as to how a drug needs to be designed and
manufactured, the cGMP regulations presents a framework for the manufacturers to develop and
follow. The most important consideration behind GMP regulations is the fact that it is necessary
to design and build quality in the product.
Figure 3 Flow chart for inspection under GMP (Aziz, 2018)
Time-period
As mentioned there are two types of trials that must be conducted before approval of a new drug.
The pre-clinical trial takes around 4 years to complete. On successful completion of pre-clinical
trial IND application is submitted to FDA. Upon approval of IND, the clinical trials commence
with phase 1, 2 and 3 and in total take 1-3 years to complete (OMNICS International, 2019). On
an average it would take around 8 to 12 years for the approval of a new drug in United States.
For generic medical product
Generic medical products are not subject to either pre-clinical or clinical trials to establish their
safety and effectiveness. This is because the safety and effectiveness of the drug compound was
already established by the new drug or the innovative biological product (of which the generic
drug is bioequivalent) through the process of NDA. Instead the manufacturer of a generic drug
has to file Abbreviated New Drug Application (ANDA). ANDA is for all generic products which
are bioequivalent or pharmaceutical equivalent of an existing product that has been approved
through the NDA process. The manufacturer or the sponsor of the generic drug provides the
required data and information through ANDA to establish that the product is bioequivalent of a
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

pre-existing innovator drug. The application should also state that the labeling and proposed use
of the drug is similar to the reference drug. However, the manufacturers of ANDA have to go
through similar inspection requirements as applied to the manufacturer of innovator drug.
of the drug is similar to the reference drug. However, the manufacturers of ANDA have to go
through similar inspection requirements as applied to the manufacturer of innovator drug.

Reference
Aziz, K. J. 2018. FDA UPDATE – The FDA’s New Drug Approval Process: Development &
Premarket Applications. Available at < https://drug-dev.com/fda-update-the-fdas-new-drug-
approval-process-development-premarket-applications/ > [Accessed 30th March 2019].
Aziz, K.J., 2017. FDA’s Design Control Requirements for Biomarkers in Drug Development.
Journal of Drug Development & Delivery, 3(1), pp.1-6.
FDA. 2019. What we do. Available at <
https://www.fda.gov/AboutFDA/WhatWeDo/default.htm > [Accessed 30th March 2019].
FDA. 2019a. New Drug Application. Available at
<https://www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/
approvalapplications/newdrugapplicationnda/default.htm> [Accessed 30th March 2019].
FDA. 2019b. Form FDA 356h. Available at <
https://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Forms/UCM082348.pdf >
[Accessed 30th March 2019].
Kaitin, K.I. and DiMasi, J.A., 2011. Pharmaceutical innovation in the 21st century: new drug
approvals in the first decade, 2000–2009. Clinical pharmacology & therapeutics, 89(2), pp.183-
188.
Kaitin, K.I., 2010. Deconstructing the drug development process: the new face of
innovation. Clinical Pharmacology & Therapeutics, 87(3), pp.356-361.
Kneller, R., 2010. The importance of new companies for drug discovery: origins of a decade of
new drugs. Nature Reviews Drug Discovery, 9(11), pp.867-882.
OMNICS International. 2019. Drug Evolution Process of IND, NDA & ANDA. Available at <
https://www.omicsonline.org/conferences-list/drug-evolution-process-of-ind-nda-anda >
[Accessed 30th March 2019].
Aziz, K. J. 2018. FDA UPDATE – The FDA’s New Drug Approval Process: Development &
Premarket Applications. Available at < https://drug-dev.com/fda-update-the-fdas-new-drug-
approval-process-development-premarket-applications/ > [Accessed 30th March 2019].
Aziz, K.J., 2017. FDA’s Design Control Requirements for Biomarkers in Drug Development.
Journal of Drug Development & Delivery, 3(1), pp.1-6.
FDA. 2019. What we do. Available at <
https://www.fda.gov/AboutFDA/WhatWeDo/default.htm > [Accessed 30th March 2019].
FDA. 2019a. New Drug Application. Available at
<https://www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/
approvalapplications/newdrugapplicationnda/default.htm> [Accessed 30th March 2019].
FDA. 2019b. Form FDA 356h. Available at <
https://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Forms/UCM082348.pdf >
[Accessed 30th March 2019].
Kaitin, K.I. and DiMasi, J.A., 2011. Pharmaceutical innovation in the 21st century: new drug
approvals in the first decade, 2000–2009. Clinical pharmacology & therapeutics, 89(2), pp.183-
188.
Kaitin, K.I., 2010. Deconstructing the drug development process: the new face of
innovation. Clinical Pharmacology & Therapeutics, 87(3), pp.356-361.
Kneller, R., 2010. The importance of new companies for drug discovery: origins of a decade of
new drugs. Nature Reviews Drug Discovery, 9(11), pp.867-882.
OMNICS International. 2019. Drug Evolution Process of IND, NDA & ANDA. Available at <
https://www.omicsonline.org/conferences-list/drug-evolution-process-of-ind-nda-anda >
[Accessed 30th March 2019].
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

The Weinberg Group. 2019. NDA submissions 505(b)(1) and 505(b)(2). Available at <
https://weinberggroup.com/regulatory-strategy-submissions/fda-submissions/505b2-pathway/ >
[Accessed 30th March 2019].
Watkins, P.B., 2011. Drug safety sciences and the bottleneck in drug development. Clinical
Pharmacology & Therapeutics, 89(6), pp.788-790.
Woodcock, J. and Woosley, R., 2008. The FDA critical path initiative and its influence on new
drug development. Annual Review Medical, 59, pp.1-12.
https://weinberggroup.com/regulatory-strategy-submissions/fda-submissions/505b2-pathway/ >
[Accessed 30th March 2019].
Watkins, P.B., 2011. Drug safety sciences and the bottleneck in drug development. Clinical
Pharmacology & Therapeutics, 89(6), pp.788-790.
Woodcock, J. and Woosley, R., 2008. The FDA critical path initiative and its influence on new
drug development. Annual Review Medical, 59, pp.1-12.
1 out of 10
Related Documents






Your All-in-One AI-Powered Toolkit for Academic Success.
+13062052269
info@desklib.com
Available 24*7 on WhatsApp / Email
![[object Object]](/_next/static/media/star-bottom.7253800d.svg)
Unlock your academic potential
Copyright © 2020–2026 A2Z Services. All Rights Reserved. Developed and managed by ZUCOL.