HSN211: Glycogen Storage Disease Dietary Handout for Parents
VerifiedAdded on 2022/09/14
|6
|1782
|21
Report
AI Summary
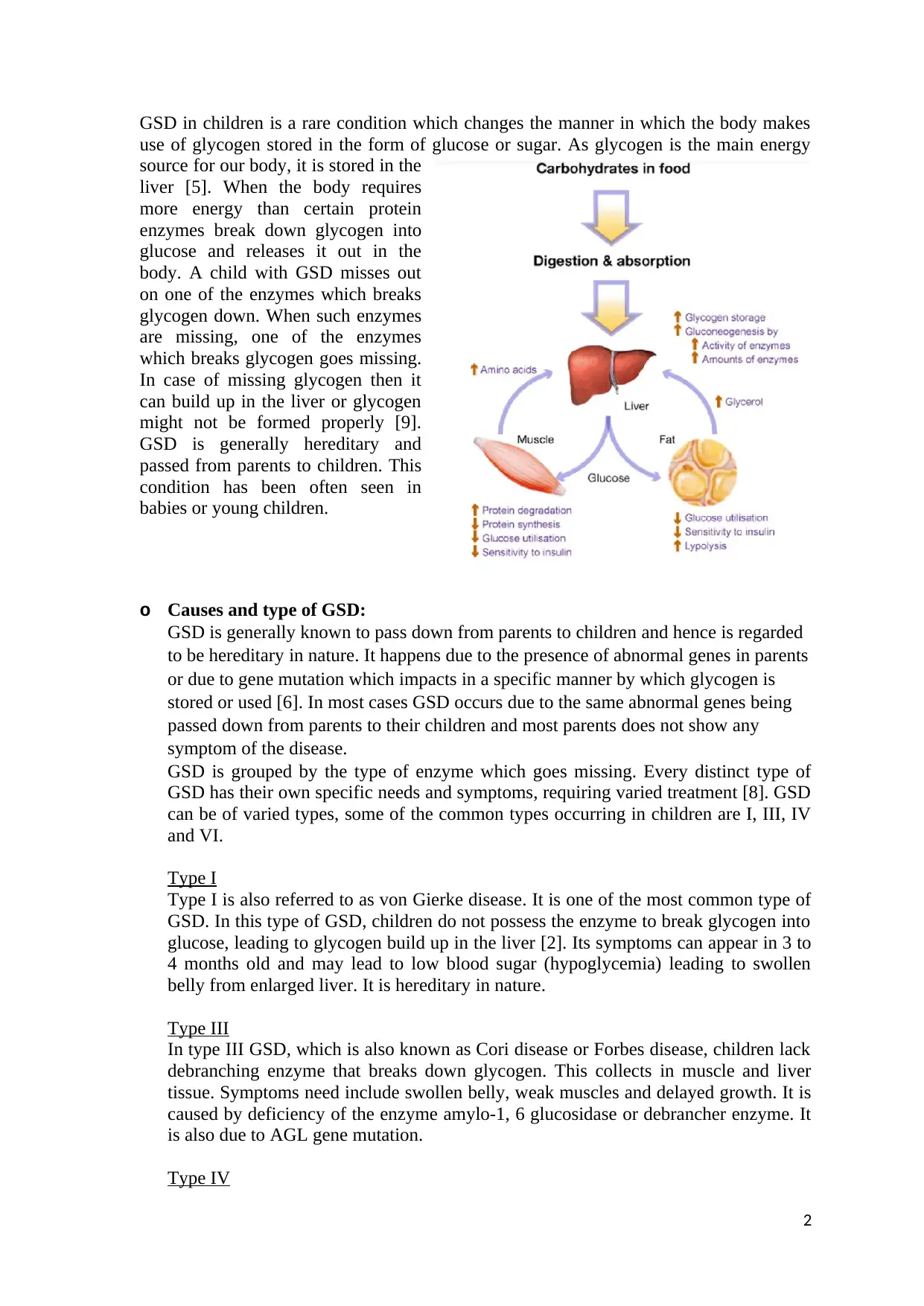
This report provides a comprehensive overview of Glycogen Storage Disease (GSD), focusing on types I, III, IV, and VI, causes, and the impact on liver function. It details the normal liver's role in glucose storage and metabolism, then explains how GSD disrupts this process. The report provides specific dietary recommendations, including the importance of complex carbohydrates, high-protein feedings, and foods to avoid for each GSD type. It emphasizes the need for a balanced diet and the importance of consulting with healthcare professionals for personalized guidance. The report is designed as a handout for parents, ensuring readability and practical application of the information, aiming to support children with GSD through proper nutrition and dietary management.




1 out of 6
Related Documents






Your All-in-One AI-Powered Toolkit for Academic Success.
+13062052269
info@desklib.com
Available 24*7 on WhatsApp / Email
![[object Object]](/_next/static/media/star-bottom.7253800d.svg)
Copyright © 2020–2026 A2Z Services. All Rights Reserved. Developed and managed by ZUCOL.