Data Structures and Algorithms: A Bioinformatics Analysis
VerifiedAdded on 2023/06/16
|9
|1285
|294
Homework Assignment
AI Summary
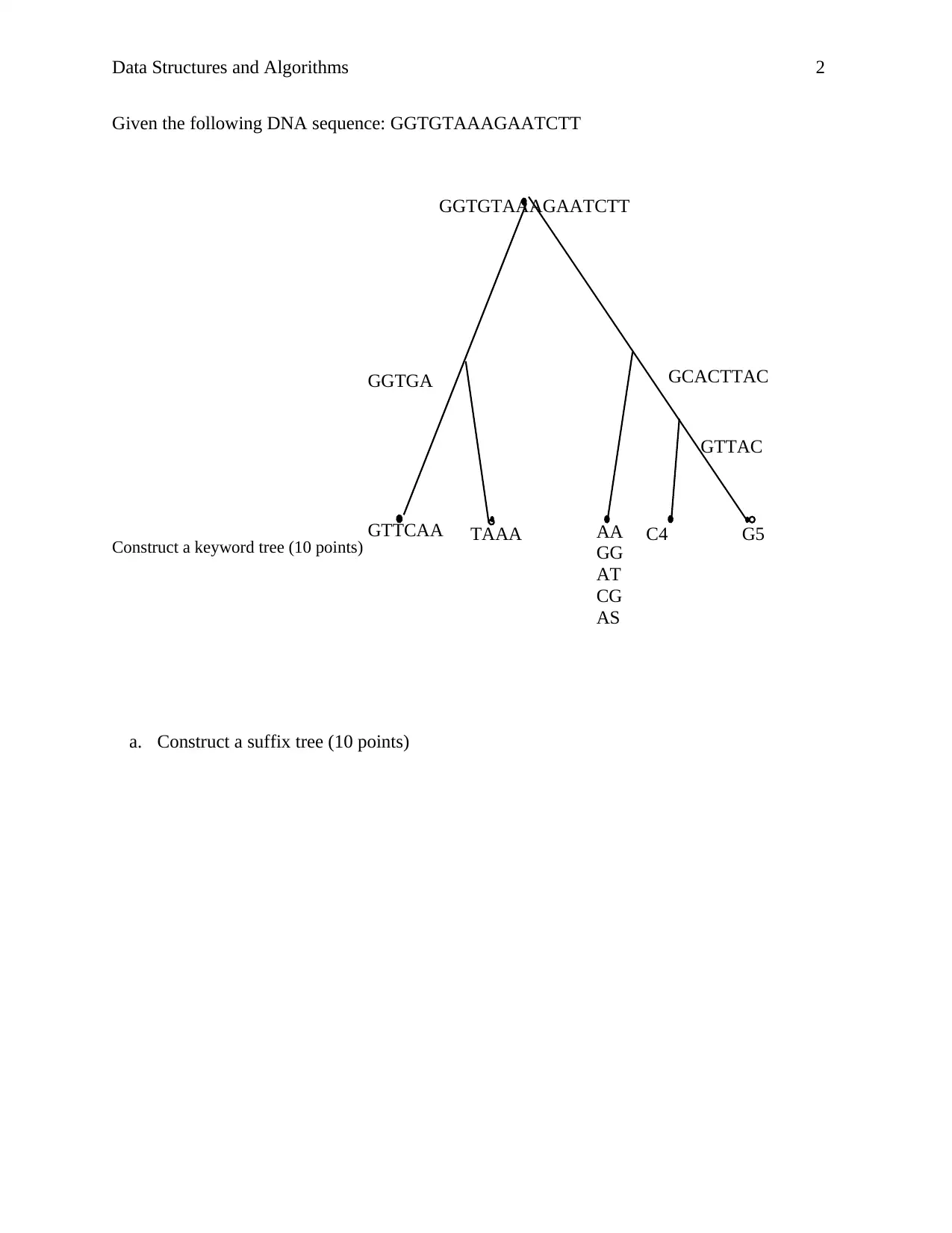
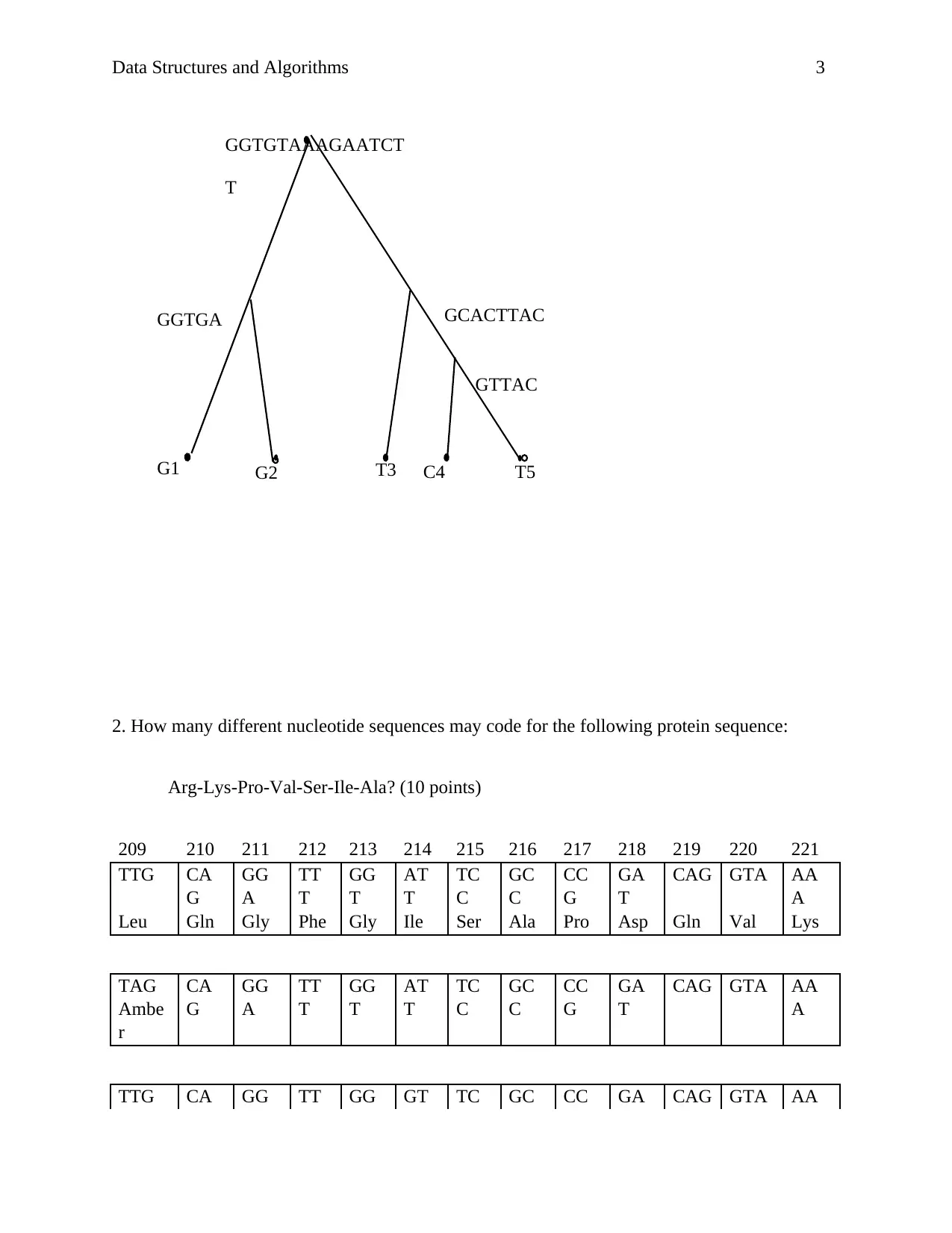
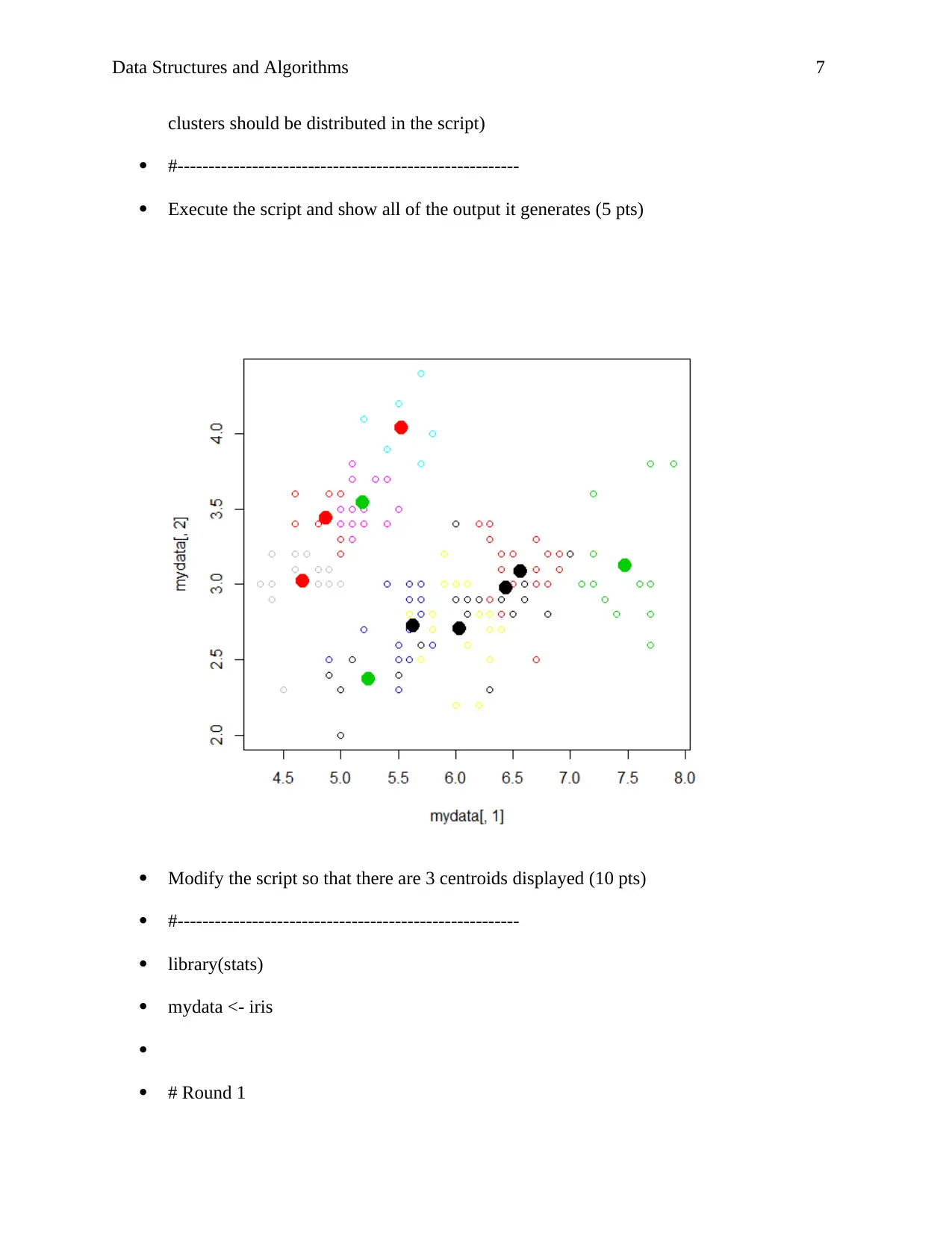
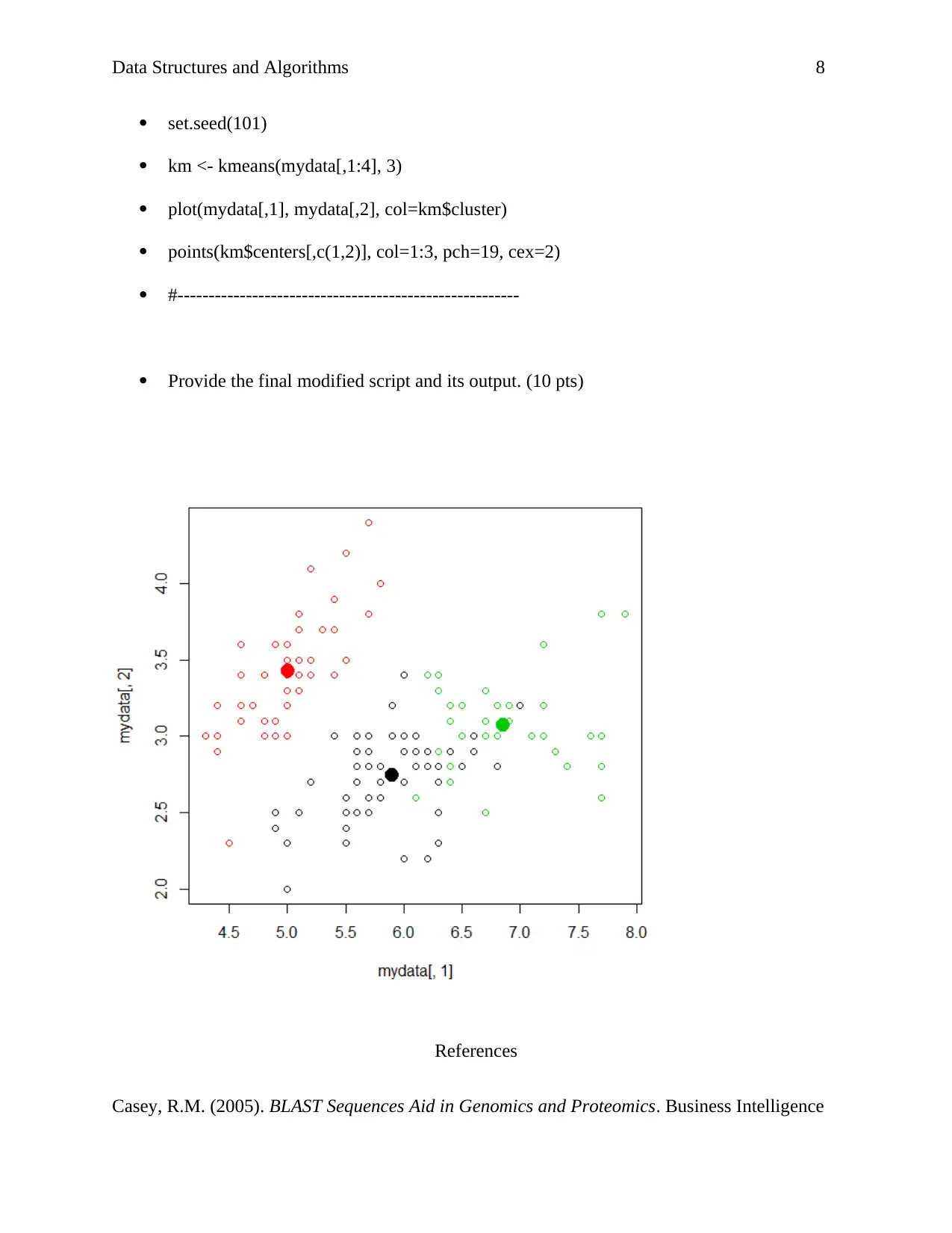
This assignment delves into various aspects of data structures and algorithms within the field of bioinformatics. It includes constructing keyword and suffix trees from a given DNA sequence, calculating the number of nucleotide sequences for a protein sequence, describing a pseudocode for identifying informative sites in a Multiple Sequence Alignment (MSA), explaining gene finding algorithms, detailing the BLAST algorithm and its statistical measures, and analyzing an R script for statistical analysis, including modifications to display a specified number of centroids. The solutions provided cover the processes and methodologies used in bioinformatics to analyze biological data and extract meaningful insights.





1 out of 9
Related Documents




Your All-in-One AI-Powered Toolkit for Academic Success.
+13062052269
info@desklib.com
Available 24*7 on WhatsApp / Email
![[object Object]](/_next/static/media/star-bottom.7253800d.svg)
Copyright © 2020–2026 A2Z Services. All Rights Reserved. Developed and managed by ZUCOL.