Bioinformatics Report: Phylogenetic Prediction and Analysis
VerifiedAdded on 2022/07/21
|5
|1299
|21
Report
AI Summary
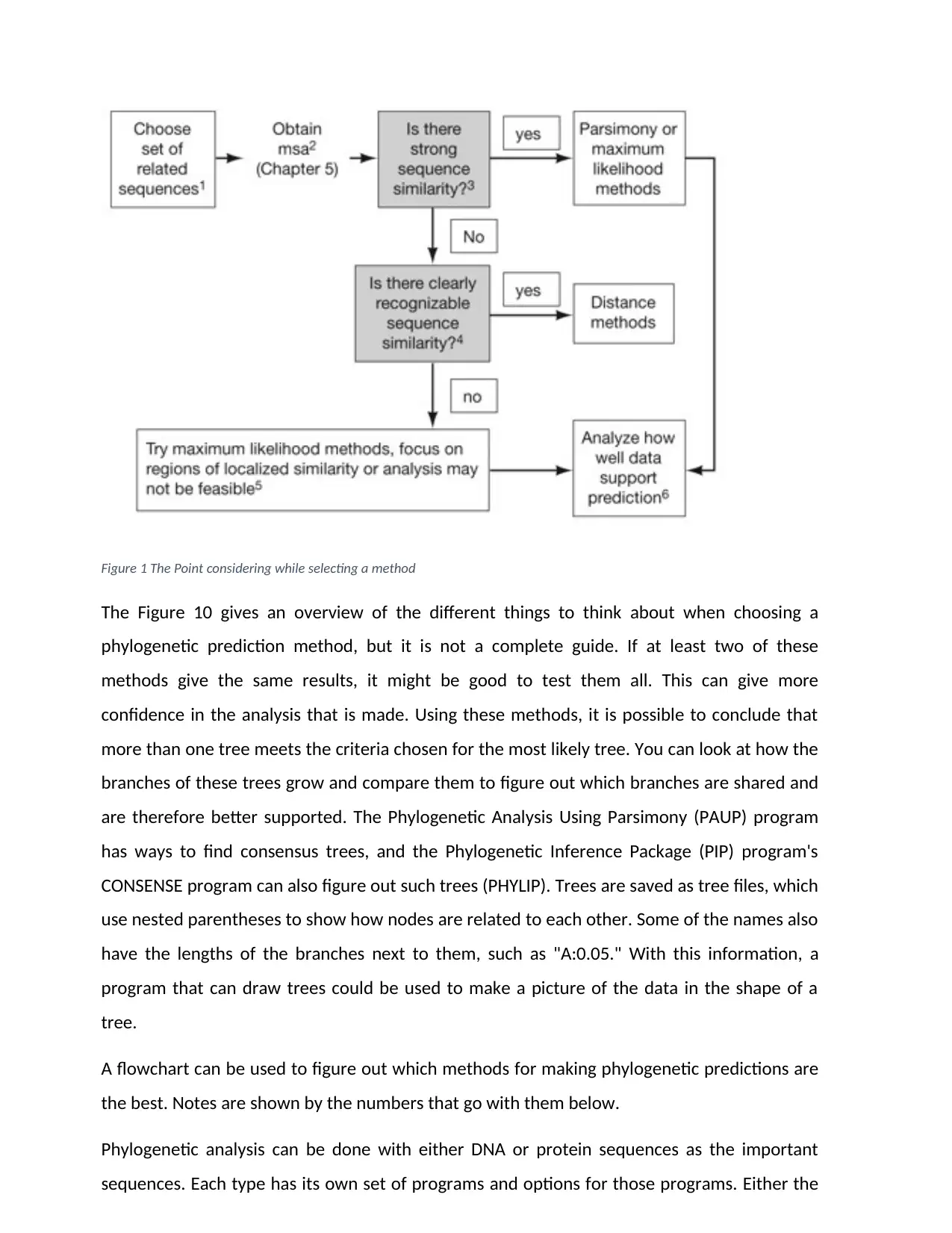
This report provides an overview of phylogenetic prediction, a crucial aspect of bioinformatics used to determine evolutionary relationships between sequences. It explores various methods for constructing evolutionary trees, including maximum parsimony, distance methods, and maximum likelihood. The report emphasizes the importance of multiple sequence alignment (MSA) and the factors to consider when selecting a suitable phylogenetic method. It explains how maximum parsimony identifies the tree with the fewest evolutionary steps, while also touching upon distance-based methods. The report also discusses the limitations of each method, such as the computational intensity of maximum likelihood and the need for sequence similarity in maximum parsimony. Overall, the document aims to guide researchers in selecting and applying the appropriate phylogenetic prediction methods for analyzing a set of related sequences.




1 out of 5
Related Documents






Your All-in-One AI-Powered Toolkit for Academic Success.
+13062052269
info@desklib.com
Available 24*7 on WhatsApp / Email
![[object Object]](/_next/static/media/star-bottom.7253800d.svg)
Copyright © 2020–2026 A2Z Services. All Rights Reserved. Developed and managed by ZUCOL.