Cystic Fibrosis: Molecular Pathophysiology, Treatment, and Questions
VerifiedAdded on 2021/04/17
|8
|1489
|158
Report
AI Summary
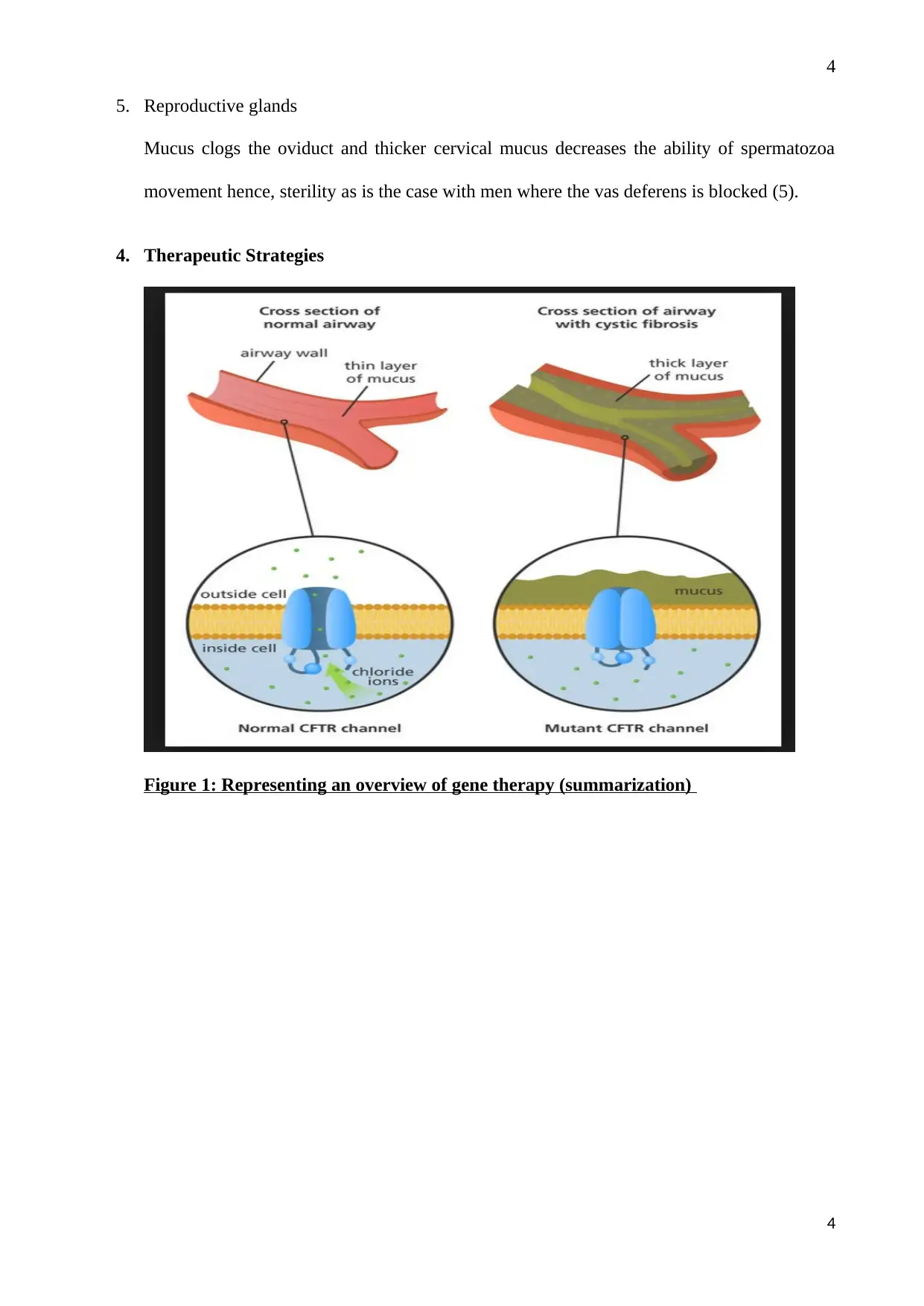
This report provides a comprehensive overview of Cystic Fibrosis (CF), an epithelial disease characterized by the overproduction of mucus due to CFTR gene mutations. It explores the 3 key features of the disease, focusing on the molecular pathophysiology, particularly the impact on the respiratory, hematopoietic, gastrointestinal, endocrine, and reproductive systems. The report details therapeutic strategies, including gene therapy and the targeting of ion channel abnormalities. Furthermore, it poses key questions about the genetic mutations and protein function of the CFTR protein, suggesting experimental designs such as meta-analysis and RFLP to understand the implications of chromosome deletions and rearrangements and the characterization of the CFTR protein. This report is contributed by a student and is available on Desklib.







1 out of 8
Related Documents






Your All-in-One AI-Powered Toolkit for Academic Success.
+13062052269
info@desklib.com
Available 24*7 on WhatsApp / Email
![[object Object]](/_next/static/media/star-bottom.7253800d.svg)
Copyright © 2020–2026 A2Z Services. All Rights Reserved. Developed and managed by ZUCOL.