Cystic Fibrosis: Pathophysiology, Treatments, and Pharmacology
VerifiedAdded on 2021/04/24
|15
|3364
|568
Report
AI Summary
This report provides a comprehensive overview of Cystic Fibrosis (CF), a genetic disease affecting multiple organ systems. It details the normal physiology and pathophysiology of CF, including its impact on the gastrointestinal tract, pancreas, hepatobiliary system, and lungs. The report explores pharmacological treatments, specifically focusing on CFTR correctors and potentiators like Orkambi, pancreatic enzymes, and antibiotics such as Aztreonam. It covers contraindications, mechanisms of action, routes of administration, and pharmacokinetics of these drugs. The report emphasizes the clinical manifestations of CF, including chronic diarrhea, malnutrition, and chronic cough, and the importance of managing the disease through various therapeutic interventions. The content is contributed by a student and available on Desklib, a platform offering AI-based study tools for students.

Running Head: CYSTIC FIBROSIS
Cystic Fibrosis
Name of Student
Institution affiliation
Cystic Fibrosis
Name of Student
Institution affiliation
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

CYSTIC FIBROSIS 2
Table of Contents
Introduction......................................................................................................................................3
Normal physiology of cystic fibrosis...............................................................................................3
Pathophysiology of cystic fibrosis (CF)..........................................................................................4
Gastrointestinal disease................................................................................................................4
Pancreatic disease........................................................................................................................5
Hepatobiliary Disease..................................................................................................................6
Lungs............................................................................................................................................7
Pharmacological treatments of CF...................................................................................................8
CFTR Correctors and Potentiators; Orkambi..............................................................................8
Contraindications......................................................................................................................8
Mode of Action........................................................................................................................8
Routes of administration..........................................................................................................9
Pancreatic enzymes......................................................................................................................9
The mechanism of action.........................................................................................................9
Pharmacokinetics of the drug...................................................................................................9
Routes of administration........................................................................................................10
Contraindications....................................................................................................................11
Antibiotics..................................................................................................................................11
Mode of Action......................................................................................................................11
Route of administration..........................................................................................................12
Pharmacokinetics....................................................................................................................12
Contraindications....................................................................................................................12
Conclusion.....................................................................................................................................13
Table of Contents
Introduction......................................................................................................................................3
Normal physiology of cystic fibrosis...............................................................................................3
Pathophysiology of cystic fibrosis (CF)..........................................................................................4
Gastrointestinal disease................................................................................................................4
Pancreatic disease........................................................................................................................5
Hepatobiliary Disease..................................................................................................................6
Lungs............................................................................................................................................7
Pharmacological treatments of CF...................................................................................................8
CFTR Correctors and Potentiators; Orkambi..............................................................................8
Contraindications......................................................................................................................8
Mode of Action........................................................................................................................8
Routes of administration..........................................................................................................9
Pancreatic enzymes......................................................................................................................9
The mechanism of action.........................................................................................................9
Pharmacokinetics of the drug...................................................................................................9
Routes of administration........................................................................................................10
Contraindications....................................................................................................................11
Antibiotics..................................................................................................................................11
Mode of Action......................................................................................................................11
Route of administration..........................................................................................................12
Pharmacokinetics....................................................................................................................12
Contraindications....................................................................................................................12
Conclusion.....................................................................................................................................13

CYSTIC FIBROSIS 3
Introduction
This is a genetic disease commonly found among the Caucasians. However, it is rare in
other races. The gene in cystic fibrosis has 230 alleles on chromosome seven. Cystic Fibrosis
Transmembrane Conductance Regulator (CFTR) is the protein encoded by the gene. The protein
functions as an ion channels. A mutation of this gene causes cystic fibrosis. The mutations affect
the CTFR protein in many ways thus leading to a dysfunctional CTFR within the apical
membrane in epithelial cells. The inability to transport ions leads to accumulation of mucus in
the epithelia of organs such as gastrointestinal tract, lungs, sweat glands and the hepatobiliary
system (Jane, 2010).
Normal physiology of cystic fibrosis
Cystic Fibrosis Transmembrane Conductance Regulator is the most vital chloride channel
in the epithelial of tissues that performs functions such as water volume absorption and
absorption of salts in lungs and sweat ducts. Moreover, it is found in the pancreas and lungs
where it secretes water. All the processes above involve transport of chloride ions. Therefore,
cystic fibrosis interrupts ion transport, which leads to several clinical effects (Jane, 2010).
Introduction
This is a genetic disease commonly found among the Caucasians. However, it is rare in
other races. The gene in cystic fibrosis has 230 alleles on chromosome seven. Cystic Fibrosis
Transmembrane Conductance Regulator (CFTR) is the protein encoded by the gene. The protein
functions as an ion channels. A mutation of this gene causes cystic fibrosis. The mutations affect
the CTFR protein in many ways thus leading to a dysfunctional CTFR within the apical
membrane in epithelial cells. The inability to transport ions leads to accumulation of mucus in
the epithelia of organs such as gastrointestinal tract, lungs, sweat glands and the hepatobiliary
system (Jane, 2010).
Normal physiology of cystic fibrosis
Cystic Fibrosis Transmembrane Conductance Regulator is the most vital chloride channel
in the epithelial of tissues that performs functions such as water volume absorption and
absorption of salts in lungs and sweat ducts. Moreover, it is found in the pancreas and lungs
where it secretes water. All the processes above involve transport of chloride ions. Therefore,
cystic fibrosis interrupts ion transport, which leads to several clinical effects (Jane, 2010).
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

CYSTIC FIBROSIS 4
Figure 1: The CTFR has two blocks of six trans-membrane domains and two nucleotide-
binding domains. Furthermore, it has a regulatory domain that has phosphorylation sites for PKA
and PKC. NBD1 participates in the formation of ion channels (Becq, 2010).
The disease manifestation in CF is changeable. The manifestation can occur in the child
stage or later stages of life. The basic symptoms are chronic diarrhea, malnutrition, and chronic
cough. Furthermore, CF affects many body organs (Jane, 2010).
Pathophysiology of cystic fibrosis (CF).
CF affects secretory granules and intracellular organs, each organ has a unique clinical
response. In addition to that, defects in the respiratory tract lead to high mortality in 90% of
patients.CF causes micro vascular and macro vascular complications caused by the deterioration
of lungs (Adriana, Aragao & Rita, 2013).
Gastrointestinal disease.
Gastrointestinal symptoms include vomiting, nausea, indigestion, and malnutrition. In
addition to that, other symptoms are esophageal reflux, cholelithiasis, and distal intestine
syndrome. Despite conventional treatments, some patients continue to have complicated
gastrointestinal problems such as chronic inflammation (Adriana, Aragao & Rita, 2013).
It is reported that eosinophilic esophasitis (EoE) can be presented in patients aged 4, 12,
and 15. Furthermore, EoE has been under appreciated due to symptom overlap with other
disorders of the intestines such as the gastro-esophageal reflux. (Adriana, Aragao & Rita, 2013).
The entire process of digestion is affected by CF thus leading to malabsoption of
nutrients, gastrointestinal problems, and malnutrition. Other signs include abdominal pains,
fibronising colonopathy and bond obstruction syndrome. The main cause of abdominal pain is
Figure 1: The CTFR has two blocks of six trans-membrane domains and two nucleotide-
binding domains. Furthermore, it has a regulatory domain that has phosphorylation sites for PKA
and PKC. NBD1 participates in the formation of ion channels (Becq, 2010).
The disease manifestation in CF is changeable. The manifestation can occur in the child
stage or later stages of life. The basic symptoms are chronic diarrhea, malnutrition, and chronic
cough. Furthermore, CF affects many body organs (Jane, 2010).
Pathophysiology of cystic fibrosis (CF).
CF affects secretory granules and intracellular organs, each organ has a unique clinical
response. In addition to that, defects in the respiratory tract lead to high mortality in 90% of
patients.CF causes micro vascular and macro vascular complications caused by the deterioration
of lungs (Adriana, Aragao & Rita, 2013).
Gastrointestinal disease.
Gastrointestinal symptoms include vomiting, nausea, indigestion, and malnutrition. In
addition to that, other symptoms are esophageal reflux, cholelithiasis, and distal intestine
syndrome. Despite conventional treatments, some patients continue to have complicated
gastrointestinal problems such as chronic inflammation (Adriana, Aragao & Rita, 2013).
It is reported that eosinophilic esophasitis (EoE) can be presented in patients aged 4, 12,
and 15. Furthermore, EoE has been under appreciated due to symptom overlap with other
disorders of the intestines such as the gastro-esophageal reflux. (Adriana, Aragao & Rita, 2013).
The entire process of digestion is affected by CF thus leading to malabsoption of
nutrients, gastrointestinal problems, and malnutrition. Other signs include abdominal pains,
fibronising colonopathy and bond obstruction syndrome. The main cause of abdominal pain is
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

CYSTIC FIBROSIS 5
gastro-esophageal reflux disease, pancreatitis, gastritis, and biliary tract disease (Stockley &
Robert, 2015).
The Distal Bowel Obstruction Syndrome affects the right colon. The syndrome is
triggered by factors such as replacement of pancreatic enzymes, dehydration, and medicines that
interfere with motility. Symptoms of the disease are decreased defecation and colic pain.
Moreover, fibronising colonopathy causes alteration of the colon submucosa, progressive
fibrosis, and inflammation. Clinical symptoms are abdominal distention after feeding, pain,
difficulty in gaining weight and anorexia (Stockley & Robert, 2015).
Pancreatic disease.
Dysfunction of CTFR mainly affects the pancreas. The pancreas produces enzymes used
in food digestion. CF leads to pancreatic insufficiency hence faecal loss of fat. Every genotype in
the mutation of CTFR is involved in the loss of pancreatic function (Adriana, Aragao & Rita,
2013).
Pancreatic exocrine insufficiency (PEI) causes intestinal malabsorption that affects
approximately 90% of patients. When PEI is inadequately treated, it leads to high energy losses
from stool, which determines malnutrition and energy imbalance (Adriana, Aragao & Rita,
2013).
PEI is characterised by difficulty in gaining weight, diarrhea, and malabsorption of
vitamins. Vitamin supplements are recommended in diet. Vitamin D is an important vitamin
since it is used in mineralization, lack of the vitamin causes depression. Most CF patients have
hypovitaminosis that is elevated by the increase in age and obesity (Proesmans, Vermeulen, &
De Boeck, 2008). The first sign of pancreatic insufficiency is the obstruction of the ileus by thick
gastro-esophageal reflux disease, pancreatitis, gastritis, and biliary tract disease (Stockley &
Robert, 2015).
The Distal Bowel Obstruction Syndrome affects the right colon. The syndrome is
triggered by factors such as replacement of pancreatic enzymes, dehydration, and medicines that
interfere with motility. Symptoms of the disease are decreased defecation and colic pain.
Moreover, fibronising colonopathy causes alteration of the colon submucosa, progressive
fibrosis, and inflammation. Clinical symptoms are abdominal distention after feeding, pain,
difficulty in gaining weight and anorexia (Stockley & Robert, 2015).
Pancreatic disease.
Dysfunction of CTFR mainly affects the pancreas. The pancreas produces enzymes used
in food digestion. CF leads to pancreatic insufficiency hence faecal loss of fat. Every genotype in
the mutation of CTFR is involved in the loss of pancreatic function (Adriana, Aragao & Rita,
2013).
Pancreatic exocrine insufficiency (PEI) causes intestinal malabsorption that affects
approximately 90% of patients. When PEI is inadequately treated, it leads to high energy losses
from stool, which determines malnutrition and energy imbalance (Adriana, Aragao & Rita,
2013).
PEI is characterised by difficulty in gaining weight, diarrhea, and malabsorption of
vitamins. Vitamin supplements are recommended in diet. Vitamin D is an important vitamin
since it is used in mineralization, lack of the vitamin causes depression. Most CF patients have
hypovitaminosis that is elevated by the increase in age and obesity (Proesmans, Vermeulen, &
De Boeck, 2008). The first sign of pancreatic insufficiency is the obstruction of the ileus by thick

CYSTIC FIBROSIS 6
meconium. Infants with meconium need urgent treatment. CF patients who manage to live longer
develop chronic manifestations. Therefore, they need to be monitored closely. Diabetes is mainly
caused by damage to the pancreas, which leads to a decrease in secretion of insulin (Adriana,
Aragao & Rita, 2013).
Hepatobiliary Disease.
Figure 2: Cystic fibrosis liver with macronodular cirrhosis (Jane, 2010).
Hepatic changes in CF are caused by defects in the CTFR protein. This leads to secretion
of thick biliary that leads to biliary fibrosis. Complications by the hepatobiliary disease include;
cirrhosis, portal hypertension, ascites, bleeding and esophageal varices. This dysfunction causes
sluggishness in bile flow. Furthermore, it causes inflammatory responses that proliferate and
activate hepatic cells, which lead to cholangitis and fibrosis (Jane, 2010).
Multilobular cirrhosis is developed in the first decade of life. Many develop hypertension
with variceal bleeding complications. When two or more conditions are present in two
examinations within 1-year period, a liver disease related to CF is defined. Elevated levels of
alkaline phosphatase, aminotransferase, gamma glutamyl transferase and ultrasound
abnormalities characterize the disease (Jane, 2010).
meconium. Infants with meconium need urgent treatment. CF patients who manage to live longer
develop chronic manifestations. Therefore, they need to be monitored closely. Diabetes is mainly
caused by damage to the pancreas, which leads to a decrease in secretion of insulin (Adriana,
Aragao & Rita, 2013).
Hepatobiliary Disease.
Figure 2: Cystic fibrosis liver with macronodular cirrhosis (Jane, 2010).
Hepatic changes in CF are caused by defects in the CTFR protein. This leads to secretion
of thick biliary that leads to biliary fibrosis. Complications by the hepatobiliary disease include;
cirrhosis, portal hypertension, ascites, bleeding and esophageal varices. This dysfunction causes
sluggishness in bile flow. Furthermore, it causes inflammatory responses that proliferate and
activate hepatic cells, which lead to cholangitis and fibrosis (Jane, 2010).
Multilobular cirrhosis is developed in the first decade of life. Many develop hypertension
with variceal bleeding complications. When two or more conditions are present in two
examinations within 1-year period, a liver disease related to CF is defined. Elevated levels of
alkaline phosphatase, aminotransferase, gamma glutamyl transferase and ultrasound
abnormalities characterize the disease (Jane, 2010).
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

CYSTIC FIBROSIS 7
CF causes infertility because of congenital malformation on the vas deferens.
Furthermore, there is an excessive loss of salt from the sweat glands. CF also causes dehydration,
ion imbalance and cardiac arrhythmiasis (Jane, 2010).
Lungs.
CF plagues the lungs with persistent infections by bacteria such as P. aeruginosa and S.
aureus. Colonization by bacteria occurs at an earlier stage, which becomes hard to manage. The
on slaughter of immune cells such as neutrophils destroys the lung tissue as it responds to
infection (Budev, 2010). The sweat ducts have a problem with salt adsorption. There is a high
conductance of Na+ and Cl-. The low permeability of water leads to hypertonic adsorption with
more salt than water .CTFR is the only pathway for conductance of anions but when the function
it is lost, sweat becomes more salty (Stockley & Robert, 2015).
Figure 3: Post mortem lung with cystic fibrosis (Adriana, Aragao & Rita, 2013).
CF causes infertility because of congenital malformation on the vas deferens.
Furthermore, there is an excessive loss of salt from the sweat glands. CF also causes dehydration,
ion imbalance and cardiac arrhythmiasis (Jane, 2010).
Lungs.
CF plagues the lungs with persistent infections by bacteria such as P. aeruginosa and S.
aureus. Colonization by bacteria occurs at an earlier stage, which becomes hard to manage. The
on slaughter of immune cells such as neutrophils destroys the lung tissue as it responds to
infection (Budev, 2010). The sweat ducts have a problem with salt adsorption. There is a high
conductance of Na+ and Cl-. The low permeability of water leads to hypertonic adsorption with
more salt than water .CTFR is the only pathway for conductance of anions but when the function
it is lost, sweat becomes more salty (Stockley & Robert, 2015).
Figure 3: Post mortem lung with cystic fibrosis (Adriana, Aragao & Rita, 2013).
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser
CYSTIC FIBROSIS 8
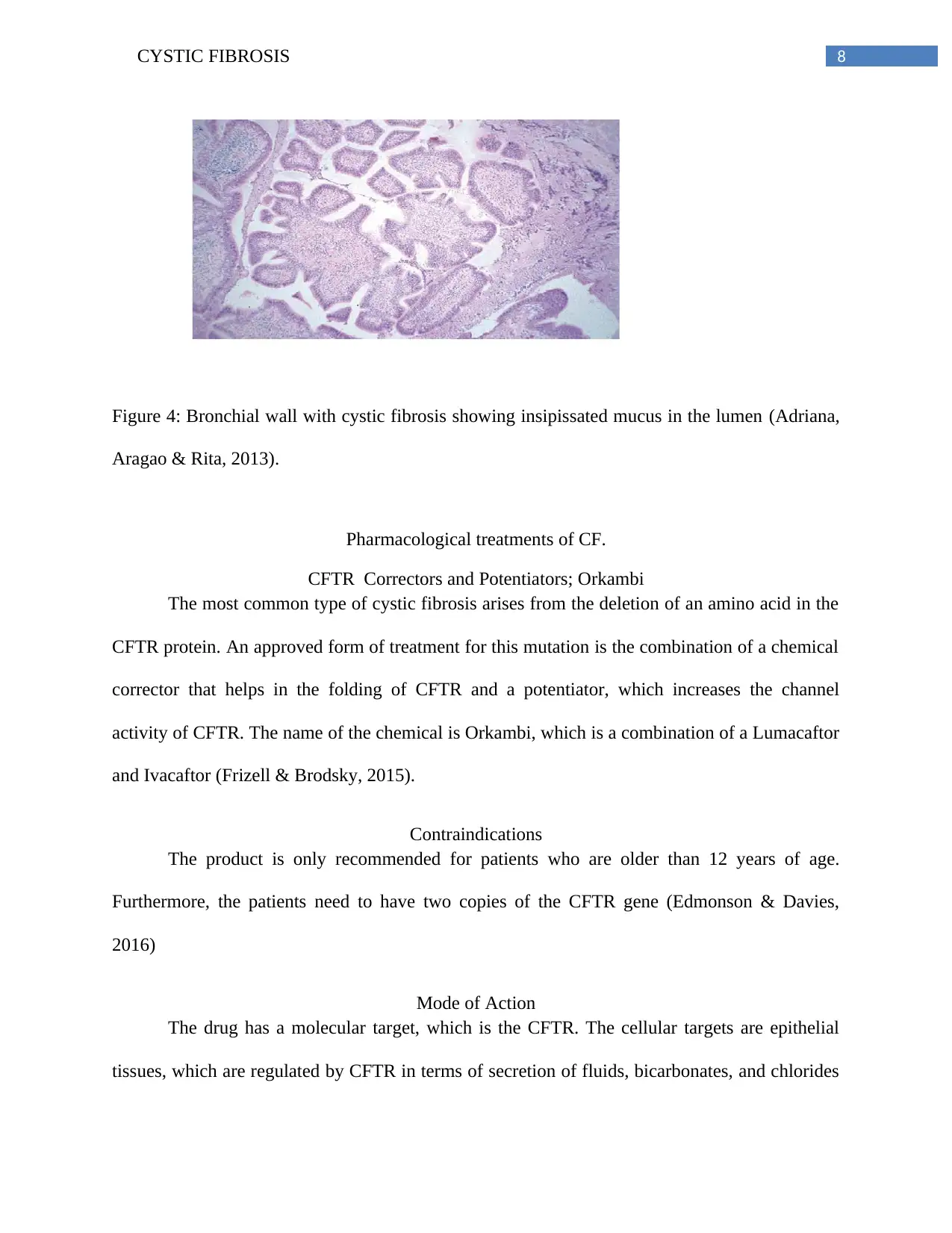
Figure 4: Bronchial wall with cystic fibrosis showing insipissated mucus in the lumen (Adriana,
Aragao & Rita, 2013).
Pharmacological treatments of CF.
CFTR Correctors and Potentiators; Orkambi
The most common type of cystic fibrosis arises from the deletion of an amino acid in the
CFTR protein. An approved form of treatment for this mutation is the combination of a chemical
corrector that helps in the folding of CFTR and a potentiator, which increases the channel
activity of CFTR. The name of the chemical is Orkambi, which is a combination of a Lumacaftor
and Ivacaftor (Frizell & Brodsky, 2015).
Contraindications
The product is only recommended for patients who are older than 12 years of age.
Furthermore, the patients need to have two copies of the CFTR gene (Edmonson & Davies,
2016)
Mode of Action
The drug has a molecular target, which is the CFTR. The cellular targets are epithelial
tissues, which are regulated by CFTR in terms of secretion of fluids, bicarbonates, and chlorides
Figure 4: Bronchial wall with cystic fibrosis showing insipissated mucus in the lumen (Adriana,
Aragao & Rita, 2013).
Pharmacological treatments of CF.
CFTR Correctors and Potentiators; Orkambi
The most common type of cystic fibrosis arises from the deletion of an amino acid in the
CFTR protein. An approved form of treatment for this mutation is the combination of a chemical
corrector that helps in the folding of CFTR and a potentiator, which increases the channel
activity of CFTR. The name of the chemical is Orkambi, which is a combination of a Lumacaftor
and Ivacaftor (Frizell & Brodsky, 2015).
Contraindications
The product is only recommended for patients who are older than 12 years of age.
Furthermore, the patients need to have two copies of the CFTR gene (Edmonson & Davies,
2016)
Mode of Action
The drug has a molecular target, which is the CFTR. The cellular targets are epithelial
tissues, which are regulated by CFTR in terms of secretion of fluids, bicarbonates, and chlorides

CYSTIC FIBROSIS 9
(Jones & Barry, 2015). Lumacaftor corrects the folding of mutant CTFR. Ivacaftor potentiates
the activity of CTFR channels. Restoration of activity and trafficking of CFTR counters defects
in secretion of fluids in intestines, sweat glands, lungs, and pancreas. The drug improves the
formation of airway surface liquid, microbe clearance, and productive mucus (Clancy & Hudock,
2017).
Routes of administration.
The drug is administered orally via the enteric administration since it is in the form of
capsules (Clancy & Hudock, 2017).
Pancreatic enzymes.
Enzyme supplements for the pancreas help in digestion when the pancreas is defective.
Most of the pancreatic enzyme supplements have different proportions of amylase, lipase, and
protease enzymes. Pancrelipase (Rx) is a pancreatic enzyme supplement also known as Pertzye,
Creon or Pancreaze (Sawczak, Getsy, Zaidi, Sun, Zaman & Gaston, 2015).
The mechanism of action.
The drug product has a combination of various enzymes such as proteases, amylase, and
lipases. Furthermore, they catalyze starch hydrolysis into short chain sugars for example maltose
and dextrins, they also catalyse hydrolysis of fats to monoglycerides, protein to peptides,
glycerol, and fatty acids in the small intestine and the duodenum (Barret, Alagely & Topol,
2012).
Pharmacokinetics of the drug.
The product is not absorbed but it acts locally within the gastro intestinal tract (Barret,
Alagely & Topol, 2012).
(Jones & Barry, 2015). Lumacaftor corrects the folding of mutant CTFR. Ivacaftor potentiates
the activity of CTFR channels. Restoration of activity and trafficking of CFTR counters defects
in secretion of fluids in intestines, sweat glands, lungs, and pancreas. The drug improves the
formation of airway surface liquid, microbe clearance, and productive mucus (Clancy & Hudock,
2017).
Routes of administration.
The drug is administered orally via the enteric administration since it is in the form of
capsules (Clancy & Hudock, 2017).
Pancreatic enzymes.
Enzyme supplements for the pancreas help in digestion when the pancreas is defective.
Most of the pancreatic enzyme supplements have different proportions of amylase, lipase, and
protease enzymes. Pancrelipase (Rx) is a pancreatic enzyme supplement also known as Pertzye,
Creon or Pancreaze (Sawczak, Getsy, Zaidi, Sun, Zaman & Gaston, 2015).
The mechanism of action.
The drug product has a combination of various enzymes such as proteases, amylase, and
lipases. Furthermore, they catalyze starch hydrolysis into short chain sugars for example maltose
and dextrins, they also catalyse hydrolysis of fats to monoglycerides, protein to peptides,
glycerol, and fatty acids in the small intestine and the duodenum (Barret, Alagely & Topol,
2012).
Pharmacokinetics of the drug.
The product is not absorbed but it acts locally within the gastro intestinal tract (Barret,
Alagely & Topol, 2012).
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

CYSTIC FIBROSIS 10
Routes of administration
Pancrelipase products are administered orally. Therapy is initiated at the lowest dose and
later increased gradually. Moreover, the product is administered individually depending on the
degree of steatorrhea, symptoms, patient response, and fat content. For children and adults, half
of the meal dose should be administered with snacks. Furthermore, the dose should be decreased
in geriatric patients because they have more weight. Therefore, they tend to digest less fat in
relation to the weight of their bodies. The capsule should be ingested with enough fluid.
Furthermore, it should not be crushed, retained in the mouth, or chewed since it causes irritation
in the oral mucosa (Marson, Bertuzzo & Ribeiro, 2015)
However, those who are unable to swallow the capsule, are advised to sprinkle contents
of the capsule on yoghurt, soft foods with a pH of less than 4.5 then ingested without chewing.
Such kind of administration is followed with fluids such as juice or water to avoid irritation on
the oral mucosa. Furthermore, it ensures that the capsules are entirely ingested and nothing is
retained in the mouth. (Kaiser, 2012).
The product can also be administered through gastrostomy tube (G-tube). A gastrostomy
tube of diameter of greater than 14 French is used to administer one or two 4000 unit capsules of
lipase with soft foods. However, if the dose needs more than 4000 –unit capsules of lipase, the
G-tube is flushed with 10 millilitres of water, and then any unused portions of the product and
soft food are discarded until the prescribed dosage is attained (Kaiser, 2012).
The product is administered to infants aged below 12 months before each feeding.
However, the capsule contents are not mixed directly into breast milk or formula because this
may reduce the efficacy of the product (Kaiser, 2012).
Routes of administration
Pancrelipase products are administered orally. Therapy is initiated at the lowest dose and
later increased gradually. Moreover, the product is administered individually depending on the
degree of steatorrhea, symptoms, patient response, and fat content. For children and adults, half
of the meal dose should be administered with snacks. Furthermore, the dose should be decreased
in geriatric patients because they have more weight. Therefore, they tend to digest less fat in
relation to the weight of their bodies. The capsule should be ingested with enough fluid.
Furthermore, it should not be crushed, retained in the mouth, or chewed since it causes irritation
in the oral mucosa (Marson, Bertuzzo & Ribeiro, 2015)
However, those who are unable to swallow the capsule, are advised to sprinkle contents
of the capsule on yoghurt, soft foods with a pH of less than 4.5 then ingested without chewing.
Such kind of administration is followed with fluids such as juice or water to avoid irritation on
the oral mucosa. Furthermore, it ensures that the capsules are entirely ingested and nothing is
retained in the mouth. (Kaiser, 2012).
The product can also be administered through gastrostomy tube (G-tube). A gastrostomy
tube of diameter of greater than 14 French is used to administer one or two 4000 unit capsules of
lipase with soft foods. However, if the dose needs more than 4000 –unit capsules of lipase, the
G-tube is flushed with 10 millilitres of water, and then any unused portions of the product and
soft food are discarded until the prescribed dosage is attained (Kaiser, 2012).
The product is administered to infants aged below 12 months before each feeding.
However, the capsule contents are not mixed directly into breast milk or formula because this
may reduce the efficacy of the product (Kaiser, 2012).
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

CYSTIC FIBROSIS 11
Contraindications.
The product should not be chewed, crushed, or retained in the mouth since this causes
irritation on the oral mucosa and reduces efficacy of the product. Furthermore, the product has
documented hypersensitivity (Gerald, 2012).
Furthermore, the maximum dosage of the product should not be exceeded because of
fibronising colonopathy. A dosage should not be more than 6000 units of lipase per kilogram per
meal since this is associated with colonic stricture, fibronising colonopathy especially in children
below 12 years. Uric acid levels in patients with hyperuricemia, renal impairment or gout should
be monitored because of hyperuricemia. Patients with porcine proteins or known allergies are
cautioned against using the product (Gerald, 2012).
Antibiotics
Antibiotics are used in the treatments of cystic fibrosis. The antibiotics are used against
microorganisms that are associated with the disease. An example of an antibiotic that used is
Aztreonam ( Rx) also known as Cayston (Murphy & Caraher, 2016).
Mode of Action.
The product inhibits synthesis of the cell wall by binding to the penicillin binding protein
3. Furthermore, the protein inhibits biosynthesis of the cell wall in members of the monobactam
family (Murphy & Caraher, 2016).
Route of administration
It improves symptoms in CF patients infected with P. aeruginosa.75 mg of the product is
inhaled for 28 days. An Alterna nebulizer is used in administration of the product; however,
inhalation should not be repeated for another 28 days after completion .Each dose of the product
should be administered within four hours. A bronchodilator is used before administration; short
Contraindications.
The product should not be chewed, crushed, or retained in the mouth since this causes
irritation on the oral mucosa and reduces efficacy of the product. Furthermore, the product has
documented hypersensitivity (Gerald, 2012).
Furthermore, the maximum dosage of the product should not be exceeded because of
fibronising colonopathy. A dosage should not be more than 6000 units of lipase per kilogram per
meal since this is associated with colonic stricture, fibronising colonopathy especially in children
below 12 years. Uric acid levels in patients with hyperuricemia, renal impairment or gout should
be monitored because of hyperuricemia. Patients with porcine proteins or known allergies are
cautioned against using the product (Gerald, 2012).
Antibiotics
Antibiotics are used in the treatments of cystic fibrosis. The antibiotics are used against
microorganisms that are associated with the disease. An example of an antibiotic that used is
Aztreonam ( Rx) also known as Cayston (Murphy & Caraher, 2016).
Mode of Action.
The product inhibits synthesis of the cell wall by binding to the penicillin binding protein
3. Furthermore, the protein inhibits biosynthesis of the cell wall in members of the monobactam
family (Murphy & Caraher, 2016).
Route of administration
It improves symptoms in CF patients infected with P. aeruginosa.75 mg of the product is
inhaled for 28 days. An Alterna nebulizer is used in administration of the product; however,
inhalation should not be repeated for another 28 days after completion .Each dose of the product
should be administered within four hours. A bronchodilator is used before administration; short

CYSTIC FIBROSIS 12
acting beta agonists are administered within 15 minutes to four hours or the long acting agonists
are administered within 30 minutes to 12 hours (Murphy & Caraher, 2016).
Pharmacokinetics.
The half-life of the drug is 1.7 hours for children and 1.7 to 2.9 hours in adults.
The product has a low systemic absorption. The mean concentration in plasma for every one
hour after a single dose is 0.59 mcg/Ml. The mean concentration in plasma for every one hour
after several doses on day 1, 14 and 28 respectively is 0.65 mcg/mL. The mean concentration of
sputum is 726 mcg/g after every single dose.The peak plasma time is approximately one hour
and the protein bound is 56 % (Murphy & Caraher, 2016).
Contraindications
The drug can cause hypersensitivity reactions in the body. Furthermore, allergic reactions
have been observed in clinical trials. Therefore, it is advisable to stop treatments incase of
allergic reactions. In addition, it can cause bronchospasm. Treatment should be stopped incase
chest tightness develops. Furthermore, FEV1 should be increased within the 28 day course of
treatment. Moreover, there is an increased risk of drug resistant bacteria in the absence of
Pseudomonas aeruginosa (Chopra, Paul, Manickan, Arownow & Maguire, 2015).
Conclusion
Cystic fibrosis is the most common disorder among the Caucasians that is relevant in
clinical practice. The lethal condition affects several organs. Furthermore, it has many clinical
problems with varying severity according to the organ. The pathophysiology of the disease is
well described. Furthermore, improved therapy and new treatments are increasing life
expectancy of CF patients. Drug products used in treatment of cystic fibrosis include CFTR
correctors and potentiators, pancreatic enzymes and antibiotics. They have various
acting beta agonists are administered within 15 minutes to four hours or the long acting agonists
are administered within 30 minutes to 12 hours (Murphy & Caraher, 2016).
Pharmacokinetics.
The half-life of the drug is 1.7 hours for children and 1.7 to 2.9 hours in adults.
The product has a low systemic absorption. The mean concentration in plasma for every one
hour after a single dose is 0.59 mcg/Ml. The mean concentration in plasma for every one hour
after several doses on day 1, 14 and 28 respectively is 0.65 mcg/mL. The mean concentration of
sputum is 726 mcg/g after every single dose.The peak plasma time is approximately one hour
and the protein bound is 56 % (Murphy & Caraher, 2016).
Contraindications
The drug can cause hypersensitivity reactions in the body. Furthermore, allergic reactions
have been observed in clinical trials. Therefore, it is advisable to stop treatments incase of
allergic reactions. In addition, it can cause bronchospasm. Treatment should be stopped incase
chest tightness develops. Furthermore, FEV1 should be increased within the 28 day course of
treatment. Moreover, there is an increased risk of drug resistant bacteria in the absence of
Pseudomonas aeruginosa (Chopra, Paul, Manickan, Arownow & Maguire, 2015).
Conclusion
Cystic fibrosis is the most common disorder among the Caucasians that is relevant in
clinical practice. The lethal condition affects several organs. Furthermore, it has many clinical
problems with varying severity according to the organ. The pathophysiology of the disease is
well described. Furthermore, improved therapy and new treatments are increasing life
expectancy of CF patients. Drug products used in treatment of cystic fibrosis include CFTR
correctors and potentiators, pancreatic enzymes and antibiotics. They have various
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide
1 out of 15
Related Documents






Your All-in-One AI-Powered Toolkit for Academic Success.
+13062052269
info@desklib.com
Available 24*7 on WhatsApp / Email
![[object Object]](/_next/static/media/star-bottom.7253800d.svg)
Unlock your academic potential
Copyright © 2020–2026 A2Z Services. All Rights Reserved. Developed and managed by ZUCOL.