Metabolic Disorder Analysis: Fumarase Deficiency Report, 2017
VerifiedAdded on 2023/06/03
|13
|3093
|456
Report
AI Summary
This report provides a comprehensive overview of Fumarase Deficiency, also known as Fumarate hydratase insufficiency or Fumaric aciduria, a genetic and congenital metabolic disorder affecting mitochondrial metabolism. It details the role of the fumarate hydratase enzyme (FH), its deficiency due to mutations in the FH gene, and its impact on the TCA cycle, leading to various health issues like Leigh syndrome and cardiomyopathy. The report explores the enzymatic reaction, the structure of fumarate hydrogenase, and the TCA cycle pathway. It includes case studies, research findings from studies conducted in Turkey and BMC medical genetics, diagnostic methods involving genetic and biochemical tests, and the absence of effective treatments, focusing on supportive care and management strategies. The report also touches upon relevant policies like the Neonatal bloodspot screening program in Australia and discusses the importance of early diagnosis and intervention.

Fumarase Deficiency 1
FUMARASE DEFICIENCY
(Student Name)
(University)
FUMARASE DEFICIENCY
(Student Name)
(University)
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

Fumarase Deficiency 2
Fumarase Deficiency
Overview
Fumarase deficiency is also referred to as Fumarate hydratase insufficiency or Fumaric aciduria.
The disorder is classified into the categories of Genetic and Congenital illnesses, Nervous system
infections and metabolic disorders (3). The disease affects a person’s mitochondrial metabolism.
The deficiency in the fumarase is as a result of fumarate hydratase catalyst impairment. The
succinate shortage in dehydrogenase distresses the mitochondrial composite II which joins the
electron conveyance chain with the TCA cycle (4). Therefore, the protein biochemical pathway
is affected by the infection; the influenced protein is Fumarate hydratase class II. Fumarase
insufficiency leads to Leigh syndrome, mental and cardiomyopathy sicknesses, motor skill
weakening and leukodystrophy. The scarcity of alpha-ketoglutarate of the dehydrogenase results
in hyperlactatemia and encephalopathy and even death in children (12).
Fumarase scarcity is instigated by the mutation present in the gene of fumarate
hydrogenase (FH). The genomic factor codes the enzymatic substances that translate fumarate to
malate within the mitochondrion. Therefore, transmutations in the genetic material obstruct the
functioning of the enzyme (9). Energy production is vital to the body cells especially during the
development of the brain. Thus, when the proteins fail to work efficiently, there is a problem in
the growth of an individual’s brain (13). This leads to several symptoms and signs of fumarase
inadequacy. The TCA pathway involves the Fumaric Hydratase class II (FumC) gene which is
usually a backup for FumA during the oxidative stress and iron limitation circumstances (7).
Another name (s): fumarase; L-malate hydro-lyase; (S)-malate hydro-lyase
Systematic name: (S)-malate hydro-lyase (fumarate-forming)
Fumarase Deficiency
Overview
Fumarase deficiency is also referred to as Fumarate hydratase insufficiency or Fumaric aciduria.
The disorder is classified into the categories of Genetic and Congenital illnesses, Nervous system
infections and metabolic disorders (3). The disease affects a person’s mitochondrial metabolism.
The deficiency in the fumarase is as a result of fumarate hydratase catalyst impairment. The
succinate shortage in dehydrogenase distresses the mitochondrial composite II which joins the
electron conveyance chain with the TCA cycle (4). Therefore, the protein biochemical pathway
is affected by the infection; the influenced protein is Fumarate hydratase class II. Fumarase
insufficiency leads to Leigh syndrome, mental and cardiomyopathy sicknesses, motor skill
weakening and leukodystrophy. The scarcity of alpha-ketoglutarate of the dehydrogenase results
in hyperlactatemia and encephalopathy and even death in children (12).
Fumarase scarcity is instigated by the mutation present in the gene of fumarate
hydrogenase (FH). The genomic factor codes the enzymatic substances that translate fumarate to
malate within the mitochondrion. Therefore, transmutations in the genetic material obstruct the
functioning of the enzyme (9). Energy production is vital to the body cells especially during the
development of the brain. Thus, when the proteins fail to work efficiently, there is a problem in
the growth of an individual’s brain (13). This leads to several symptoms and signs of fumarase
inadequacy. The TCA pathway involves the Fumaric Hydratase class II (FumC) gene which is
usually a backup for FumA during the oxidative stress and iron limitation circumstances (7).
Another name (s): fumarase; L-malate hydro-lyase; (S)-malate hydro-lyase
Systematic name: (S)-malate hydro-lyase (fumarate-forming)

Fumarase Deficiency 3
The metabolic manipulator of fumarase shortage is fumarate hydratase, whose other
names are (S)-malate hydro-lyase, L-malate hydro-lyase, or fumarase. The enzyme assignment
number is of the manipulator is EC: 4.2.1.2. The TCA cycle is usually involved in the process of
FH enzyme production (1). Therefore, FumC which is said to be the backup catalyst for FumA
during the oxidative stress conditions is involved in the TCA biochemical pathways. The
enzymatic reaction consists of the conversion of fumarate substrates to L-malate (14).
Reaction: H2O +fumarate = (S)-malate
The structure of the fumarate hydrogenase manipulator
Figure 1: The structure comprises of two sites for substrate-binding which are the non-
enzymatic B site and the enzymatic site A. The locations are essential in the transportation of
products or substrates between the solvent and the active spot. The B site also links the effectors
of the allosteric substitutes (12).
Source; Bardella et al. (1)
The process is regulated by S-2,3-dicarboxyaziridine, ATP and citrate.
The metabolic manipulator of fumarase shortage is fumarate hydratase, whose other
names are (S)-malate hydro-lyase, L-malate hydro-lyase, or fumarase. The enzyme assignment
number is of the manipulator is EC: 4.2.1.2. The TCA cycle is usually involved in the process of
FH enzyme production (1). Therefore, FumC which is said to be the backup catalyst for FumA
during the oxidative stress conditions is involved in the TCA biochemical pathways. The
enzymatic reaction consists of the conversion of fumarate substrates to L-malate (14).
Reaction: H2O +fumarate = (S)-malate
The structure of the fumarate hydrogenase manipulator
Figure 1: The structure comprises of two sites for substrate-binding which are the non-
enzymatic B site and the enzymatic site A. The locations are essential in the transportation of
products or substrates between the solvent and the active spot. The B site also links the effectors
of the allosteric substitutes (12).
Source; Bardella et al. (1)
The process is regulated by S-2,3-dicarboxyaziridine, ATP and citrate.
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide
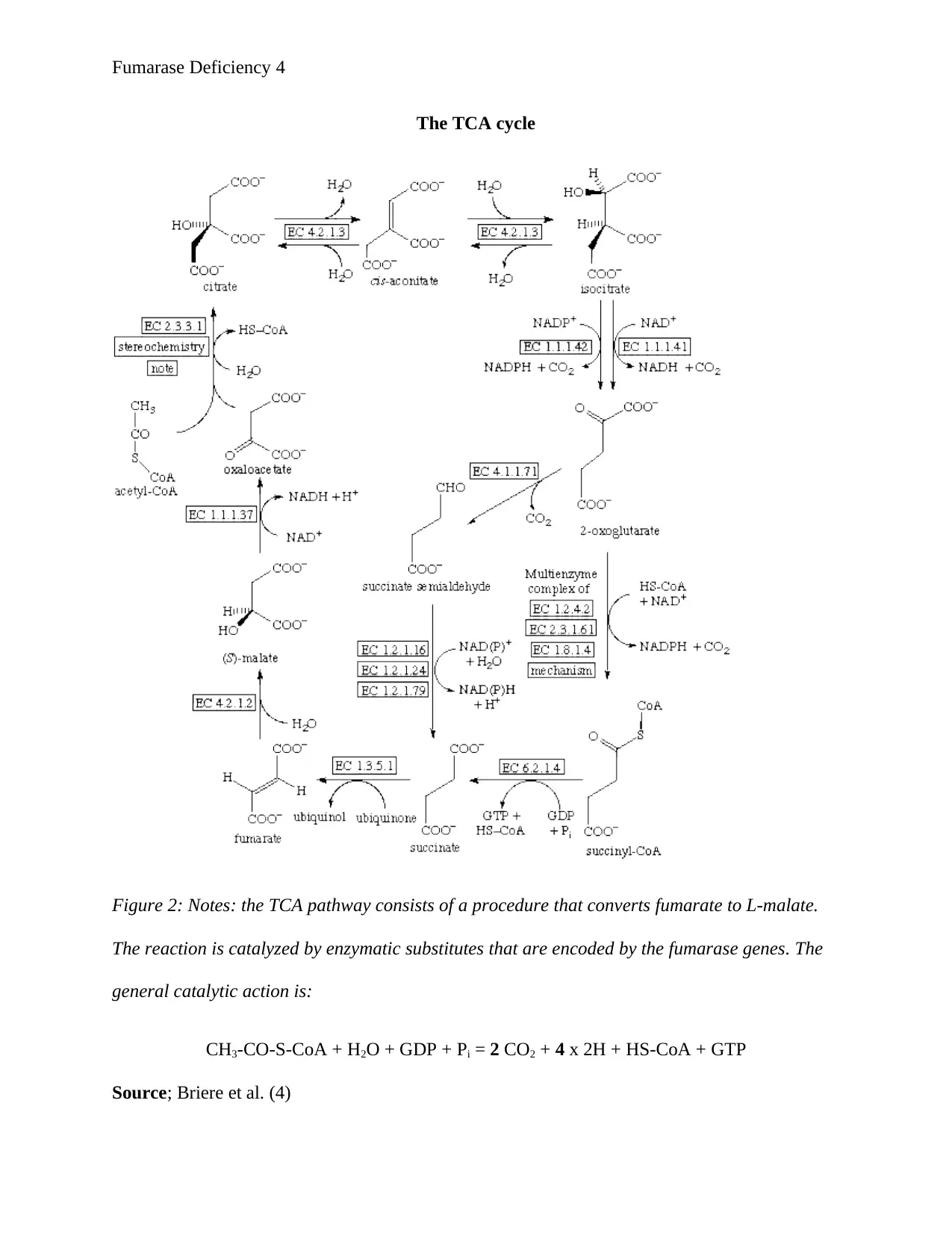
Fumarase Deficiency 4
The TCA cycle
Figure 2: Notes: the TCA pathway consists of a procedure that converts fumarate to L-malate.
The reaction is catalyzed by enzymatic substitutes that are encoded by the fumarase genes. The
general catalytic action is:
CH3-CO-S-CoA + H2O + GDP + Pi = 2 CO2 + 4 x 2H + HS-CoA + GTP
Source; Briere et al. (4)
The TCA cycle
Figure 2: Notes: the TCA pathway consists of a procedure that converts fumarate to L-malate.
The reaction is catalyzed by enzymatic substitutes that are encoded by the fumarase genes. The
general catalytic action is:
CH3-CO-S-CoA + H2O + GDP + Pi = 2 CO2 + 4 x 2H + HS-CoA + GTP
Source; Briere et al. (4)
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

Fumarase Deficiency 5
Research
In 2014, Baştuğ et al. (2) conducted an investigation in Turkey on ‘A rare cause of opistotonus;
fumaric aciduria’ was published in the Turkish Archives of Pediatrics. The research reported a
case of a male newborn in a clinic outside the Kayseri province of Turkey. The situation was
worth publishing because there had been no similar scenario of fumaric aciduria in Turkey.
Physical assessment of the baby indicated the presence of respiration difficulty accompanied
with decreased lung sounds (1). The infant was incubated, and his ventilation was mechanically
started. Bicarbonate and single surfactant dose treatment were administered as the metabolic
acidosis happened thrice after the eighth day after birth. Intravenous dexamethasone and oral
vitamin A were also provided to the child to manage the chronic lung infections. On the thirty-
eighth day, metabolic acidosis administered to the patient (16). At the forty-fifth day, the neonate
had subcostal-intercostal reactions, tachypnea, opistotonus posture, and reduced subcutaneous
lipid tissue. At day forty-five he had normal blood gases and biochemical values. However, the
amount of ammonia was higher than the standard level (7). This indicated that there was
excretion of metylmal, succinic acid, adipic acid, 4-OH phenylacetic acid, glutaric acid and
glycolic acid (16). The cranial magnetic imaging indicated the presence of polymicrogyria and
the enlargement of the lateral ventricles (11).
The cranial image
Research
In 2014, Baştuğ et al. (2) conducted an investigation in Turkey on ‘A rare cause of opistotonus;
fumaric aciduria’ was published in the Turkish Archives of Pediatrics. The research reported a
case of a male newborn in a clinic outside the Kayseri province of Turkey. The situation was
worth publishing because there had been no similar scenario of fumaric aciduria in Turkey.
Physical assessment of the baby indicated the presence of respiration difficulty accompanied
with decreased lung sounds (1). The infant was incubated, and his ventilation was mechanically
started. Bicarbonate and single surfactant dose treatment were administered as the metabolic
acidosis happened thrice after the eighth day after birth. Intravenous dexamethasone and oral
vitamin A were also provided to the child to manage the chronic lung infections. On the thirty-
eighth day, metabolic acidosis administered to the patient (16). At the forty-fifth day, the neonate
had subcostal-intercostal reactions, tachypnea, opistotonus posture, and reduced subcutaneous
lipid tissue. At day forty-five he had normal blood gases and biochemical values. However, the
amount of ammonia was higher than the standard level (7). This indicated that there was
excretion of metylmal, succinic acid, adipic acid, 4-OH phenylacetic acid, glutaric acid and
glycolic acid (16). The cranial magnetic imaging indicated the presence of polymicrogyria and
the enlargement of the lateral ventricles (11).
The cranial image
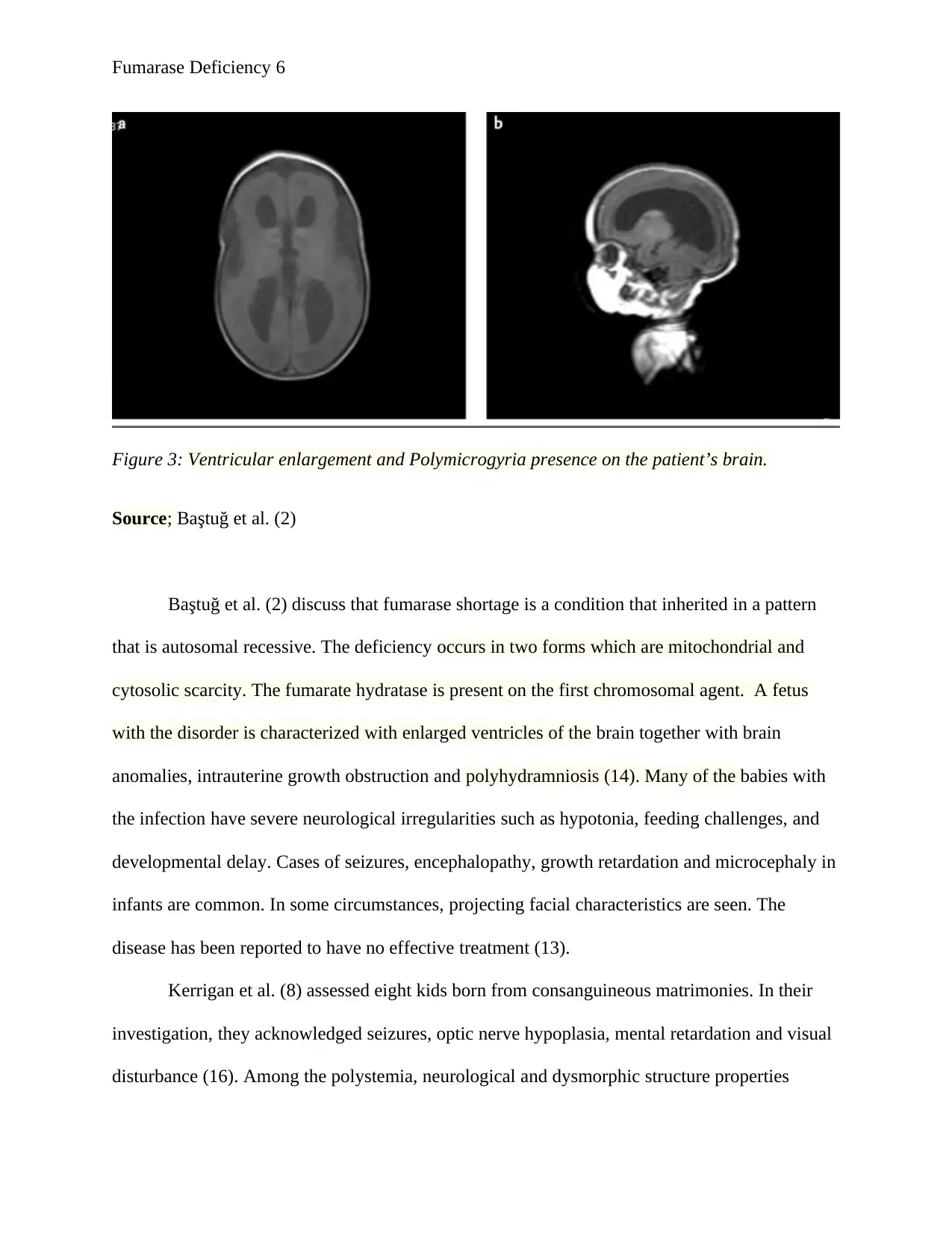
Fumarase Deficiency 6
Figure 3: Ventricular enlargement and Polymicrogyria presence on the patient’s brain.
Source; Baştuğ et al. (2)
Baştuğ et al. (2) discuss that fumarase shortage is a condition that inherited in a pattern
that is autosomal recessive. The deficiency occurs in two forms which are mitochondrial and
cytosolic scarcity. The fumarate hydratase is present on the first chromosomal agent. A fetus
with the disorder is characterized with enlarged ventricles of the brain together with brain
anomalies, intrauterine growth obstruction and polyhydramniosis (14). Many of the babies with
the infection have severe neurological irregularities such as hypotonia, feeding challenges, and
developmental delay. Cases of seizures, encephalopathy, growth retardation and microcephaly in
infants are common. In some circumstances, projecting facial characteristics are seen. The
disease has been reported to have no effective treatment (13).
Kerrigan et al. (8) assessed eight kids born from consanguineous matrimonies. In their
investigation, they acknowledged seizures, optic nerve hypoplasia, mental retardation and visual
disturbance (16). Among the polystemia, neurological and dysmorphic structure properties
Figure 3: Ventricular enlargement and Polymicrogyria presence on the patient’s brain.
Source; Baştuğ et al. (2)
Baştuğ et al. (2) discuss that fumarase shortage is a condition that inherited in a pattern
that is autosomal recessive. The deficiency occurs in two forms which are mitochondrial and
cytosolic scarcity. The fumarate hydratase is present on the first chromosomal agent. A fetus
with the disorder is characterized with enlarged ventricles of the brain together with brain
anomalies, intrauterine growth obstruction and polyhydramniosis (14). Many of the babies with
the infection have severe neurological irregularities such as hypotonia, feeding challenges, and
developmental delay. Cases of seizures, encephalopathy, growth retardation and microcephaly in
infants are common. In some circumstances, projecting facial characteristics are seen. The
disease has been reported to have no effective treatment (13).
Kerrigan et al. (8) assessed eight kids born from consanguineous matrimonies. In their
investigation, they acknowledged seizures, optic nerve hypoplasia, mental retardation and visual
disturbance (16). Among the polystemia, neurological and dysmorphic structure properties
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

Fumarase Deficiency 7
present at the neonatal stage, microcephaly and growth retardation were consistent in newborns
with fumaric aciduria. The enlargement of ventricles and polymicrogyria seen in the study of
Kerrigan et al. (8) was also observed in the research of Baştuğ et al. (2). However, Kerrigans’
analysis defined hypotonicity present in the patients, this wasn’t discussed in Baştuğs’
exploration (14).
In 2017, Chan et al. (5) investigated BMC medical genetics about Cascade Fumarate
Hydratase mutation and how it enables early identification of tumor of the kidney. The essay
analyzed a case of a child born from a family of healthy parents that were unrelated. The study
was relevant because Cascade FH mutation testing allows early diagnosis and even formulation
of life-saving treatments of renal and kidney cancers that are known to cause more than ten
thousand deaths per year in the world. Antenatal ultrasound testing identified that the girl had
increased cerebral ventricles (12). The delivery of the baby was smooth, and she had a standard
birth weight. However, she had an initial jaundice problem, but no precise treatment was needed.
At the age of three months, she illustrated symptoms of hypotonia and seizers (7). Control of
seizure became difficult with the reduced time of adequate regulatory measures to be effected
with the numerous anticonvulsant agents. On evaluation, the child was found to be
plagiocephalic with an irregular deviating strabismus, a large mouth with an upper lip that was
tented and some facial roughening (13). The MRI tests indicated the presence of deprived white
material bulk and delays in myelination. Gas-chromatography analysis of the uric acid of the
patient illustrated increased fumarate levels (4790 μmol/mmol creatinine) with very mild 2-
oxoglutarate and succinate levels. Enzyme functionality assessment showed a 20% regulation of
the fumarate hydratase actions (8). The genetic evaluation of the FH genetic factor illustrated
present at the neonatal stage, microcephaly and growth retardation were consistent in newborns
with fumaric aciduria. The enlargement of ventricles and polymicrogyria seen in the study of
Kerrigan et al. (8) was also observed in the research of Baştuğ et al. (2). However, Kerrigans’
analysis defined hypotonicity present in the patients, this wasn’t discussed in Baştuğs’
exploration (14).
In 2017, Chan et al. (5) investigated BMC medical genetics about Cascade Fumarate
Hydratase mutation and how it enables early identification of tumor of the kidney. The essay
analyzed a case of a child born from a family of healthy parents that were unrelated. The study
was relevant because Cascade FH mutation testing allows early diagnosis and even formulation
of life-saving treatments of renal and kidney cancers that are known to cause more than ten
thousand deaths per year in the world. Antenatal ultrasound testing identified that the girl had
increased cerebral ventricles (12). The delivery of the baby was smooth, and she had a standard
birth weight. However, she had an initial jaundice problem, but no precise treatment was needed.
At the age of three months, she illustrated symptoms of hypotonia and seizers (7). Control of
seizure became difficult with the reduced time of adequate regulatory measures to be effected
with the numerous anticonvulsant agents. On evaluation, the child was found to be
plagiocephalic with an irregular deviating strabismus, a large mouth with an upper lip that was
tented and some facial roughening (13). The MRI tests indicated the presence of deprived white
material bulk and delays in myelination. Gas-chromatography analysis of the uric acid of the
patient illustrated increased fumarate levels (4790 μmol/mmol creatinine) with very mild 2-
oxoglutarate and succinate levels. Enzyme functionality assessment showed a 20% regulation of
the fumarate hydratase actions (8). The genetic evaluation of the FH genetic factor illustrated
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

Fumarase Deficiency 8
complex heterozygosity for the mutation activities (hereditary from the child’s mother). The
process confirmed analysis of fumarase shortage. The growth of the girl was retarded (11).
Chan et al. (5) explain that 3-5% of the kidney tumors are genetic. Heterozygous
mutagenic occurrences in genetic materials such as the Fumarate hydratase (FH) are linked with
various renal cancer forms. The loss of somatic cells of the subsequent allele affects the kidney
typically and is associated with renal injuries (11). The heterozygous loss-of-
function FH alteration is connected to heritable leiomyomatosis and renal cell tumor (HLRCC)
disorder. HLRCC-linked RCCs are a set of histologically diverse cancers defined as cystic,
tubulocystic, papillary, cribriform or solid (6). Immuno-chemistry reveals the lack of the FH
protein manifestation with increased levels of 2SC in the cancerous growths; this is essential in
promoting the screening of the genetic makeup of the affected people. It is significant to
distinguish FH-insufficient RCC from the normal renal tissue tumors as the FH-shortage RCC
have an increased chance of early metastasis and invasion (9).
Diagnosis
Diagnosis of Fumaric aciduria involves the analysis of the patient’s symptoms, medical history,
laboratory tests, and physical fitness. Various resources are put forward for testing the disorder in
people such as the Genetic Testing Registry (GRT). It comprises of the genetic testing process
and provides the genetic data about fumarate hydratase insufficiency (16). A genomic valuation
is a form of medical examination that discovers protein, chromosome and gene transformations.
The results from the assessment rule out or confirm a suspected heritable disorder (7). The
outcomes also help in determining the people’s chances of having or escaping a genetic
infection. Various methodologies have been applied in the analysis of congenital disorders such
as the molecular genomic tests which analyze short lengthed DNA substances or single genes to
complex heterozygosity for the mutation activities (hereditary from the child’s mother). The
process confirmed analysis of fumarase shortage. The growth of the girl was retarded (11).
Chan et al. (5) explain that 3-5% of the kidney tumors are genetic. Heterozygous
mutagenic occurrences in genetic materials such as the Fumarate hydratase (FH) are linked with
various renal cancer forms. The loss of somatic cells of the subsequent allele affects the kidney
typically and is associated with renal injuries (11). The heterozygous loss-of-
function FH alteration is connected to heritable leiomyomatosis and renal cell tumor (HLRCC)
disorder. HLRCC-linked RCCs are a set of histologically diverse cancers defined as cystic,
tubulocystic, papillary, cribriform or solid (6). Immuno-chemistry reveals the lack of the FH
protein manifestation with increased levels of 2SC in the cancerous growths; this is essential in
promoting the screening of the genetic makeup of the affected people. It is significant to
distinguish FH-insufficient RCC from the normal renal tissue tumors as the FH-shortage RCC
have an increased chance of early metastasis and invasion (9).
Diagnosis
Diagnosis of Fumaric aciduria involves the analysis of the patient’s symptoms, medical history,
laboratory tests, and physical fitness. Various resources are put forward for testing the disorder in
people such as the Genetic Testing Registry (GRT). It comprises of the genetic testing process
and provides the genetic data about fumarate hydratase insufficiency (16). A genomic valuation
is a form of medical examination that discovers protein, chromosome and gene transformations.
The results from the assessment rule out or confirm a suspected heritable disorder (7). The
outcomes also help in determining the people’s chances of having or escaping a genetic
infection. Various methodologies have been applied in the analysis of congenital disorders such
as the molecular genomic tests which analyze short lengthed DNA substances or single genes to

Fumarase Deficiency 9
find mutations or variations that result in hereditary conditions like the fumarase scarcity
infections. Biochemical, genetic investigations that examine the activity level or some protein
abnormalities, the assessments aim at indicating alterations to the DNA molecule that leads to
fumarate hydratase deficiency. Chromosomal tests explore the long length DNA molecules or
the entire chromosome to identify significant genetic variations that result in fumaric aciduria.
Diagnosis also involves other management and diagnostic resources such as the testing
for Fumarate Hydratase Deficiency (12). Analysis of fumarase in patients is very vital, the
diagnosis of the disease is affirmed by the discovery of scarce activities of the enzymes of
fumarate hydratase within the white blood cells, fibroblasts or the lymphoblasts (3). The actions
of Fumarate hydratase in affected people are usually below 10% of the standard mean. However,
the residual (S)-malate hydro-lyase happenings in some affected individuals can vary between
35%-11% of the standardized average (6).
Treatment
There is no operative treatment available for the fumaric aciduria disorder. The goal of most
management systems is to enhance symptom identification techniques, increase life’s quality and
avoid complications (14). The examples of regulation strategies put forward for an active living
are the establishment of normalized anticonvulsant seizures medications. Development of
facilities for the provision of specialized services to affected persons and engagement of a
nourishing tube if required for prevention of aspiration and optimum nutrition (9). There is also
the involvement of supportive measures for treatment such as conventional therapies to regulate
seizures in the affected people (12). Gastrostomy placement is another appropriate approach for
the optimization of nutrition and prevention of aspiration. Orthopedic management and physical
psychoanalysis to avoid scoliosis and reduce contractures (6).
find mutations or variations that result in hereditary conditions like the fumarase scarcity
infections. Biochemical, genetic investigations that examine the activity level or some protein
abnormalities, the assessments aim at indicating alterations to the DNA molecule that leads to
fumarate hydratase deficiency. Chromosomal tests explore the long length DNA molecules or
the entire chromosome to identify significant genetic variations that result in fumaric aciduria.
Diagnosis also involves other management and diagnostic resources such as the testing
for Fumarate Hydratase Deficiency (12). Analysis of fumarase in patients is very vital, the
diagnosis of the disease is affirmed by the discovery of scarce activities of the enzymes of
fumarate hydratase within the white blood cells, fibroblasts or the lymphoblasts (3). The actions
of Fumarate hydratase in affected people are usually below 10% of the standard mean. However,
the residual (S)-malate hydro-lyase happenings in some affected individuals can vary between
35%-11% of the standardized average (6).
Treatment
There is no operative treatment available for the fumaric aciduria disorder. The goal of most
management systems is to enhance symptom identification techniques, increase life’s quality and
avoid complications (14). The examples of regulation strategies put forward for an active living
are the establishment of normalized anticonvulsant seizures medications. Development of
facilities for the provision of specialized services to affected persons and engagement of a
nourishing tube if required for prevention of aspiration and optimum nutrition (9). There is also
the involvement of supportive measures for treatment such as conventional therapies to regulate
seizures in the affected people (12). Gastrostomy placement is another appropriate approach for
the optimization of nutrition and prevention of aspiration. Orthopedic management and physical
psychoanalysis to avoid scoliosis and reduce contractures (6).
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

Fumarase Deficiency 10
Policies
The Neonatal bloodspot screening (NBS) program outline for Australia aims at the identification
of rare non-genomic and hereditary illnesses in children immediately after delivery (15). The
plan is essential as it gives the medical professionals ample time to begin the therapeutic
activities that evade the growth of irreversible, progressive and severe disabilities (10).
International NBS stratagems are applied in various nations to attain the targeted concern of
maintaining a healthy environment (15).
The appraisal of the Present Intercontinental Decision-Making Procedures for the
Newborn Screening is another policy put forward regarding the fumarase insufficiency infections
(10). The operation of the Neonatal bloodspot screening plan has been effective in Australia and
other countries around the world (15). However, with the recent technological advancements in
the medical fields, there is a need for new strategies for the screening outlines. Most countries
have realized the necessity of ensuring an evidence-based resolution making and transparent
system for the provision of safe and quality health care to the patients (10).
Works cited
Policies
The Neonatal bloodspot screening (NBS) program outline for Australia aims at the identification
of rare non-genomic and hereditary illnesses in children immediately after delivery (15). The
plan is essential as it gives the medical professionals ample time to begin the therapeutic
activities that evade the growth of irreversible, progressive and severe disabilities (10).
International NBS stratagems are applied in various nations to attain the targeted concern of
maintaining a healthy environment (15).
The appraisal of the Present Intercontinental Decision-Making Procedures for the
Newborn Screening is another policy put forward regarding the fumarase insufficiency infections
(10). The operation of the Neonatal bloodspot screening plan has been effective in Australia and
other countries around the world (15). However, with the recent technological advancements in
the medical fields, there is a need for new strategies for the screening outlines. Most countries
have realized the necessity of ensuring an evidence-based resolution making and transparent
system for the provision of safe and quality health care to the patients (10).
Works cited
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

Fumarase Deficiency 11
1. Bardella C, El‐Bahrawy M, Frizzell N, Adam J, Ternette N, Hatipoglu E, Howarth K,
O'flaherty L, Roberts I, Turner G, Taylor J. Aberrant succination of proteins in
fumarate hydratase‐deficient mice and HLRCC patients is a robust biomarker of
mutation status. The Journal of pathology. 2016; 225(1):4-11.
2. Baştuğ O, Kardaş F, Öztürk MA, Halis H, Memur Ş, Korkmaz L, Tağ Z, Güneş T. A
rare cause of opistotonus; fumaric aciduria: The first case presentation in Turkey.
Turkish Archives of Pediatrics/Türk Pediatri Arşivi. 2014; 49(1):74.
3. Bailey JP, Launonen V, Tomlinson IP. The FH mutation database: an online database
of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome
and congenital fumarase deficiency. BMC medical genetics. 2016; 9(1):20.
4. Briere JJ, Favier J, Gimenez-Roqueplo AP, Rustin P. Tricarboxylic acid cycle
dysfunction as a cause of human diseases and tumor formation. American Journal of
Physiology-cell physiology. 2016; 291(6): C1114-20.
5. Chan MM, Barnicoat A, Mumtaz F, Aitchison M, Side L, Brittain H, Bates AW, Gale
DP. Cascade Fumarate Hydratase mutation screening allows early detection of kidney
tumour: a case report. BMC medical genetics. 2017; 18(1):79.
6. Frezza C, Zheng L, Folger O, Rajagopalan KN, MacKenzie ED, Jerby L, Micaroni
M, Chaneton B, Adam J, Hedley A, Kalna G. Haem oxygenase is synthetically lethal
with the tumour suppressor fumarate hydratase. Nature. 2015; 477(7363):225.
7. Joseph NM, Solomon DA, Frizzell N, Rabban JT, Zaloudek C, Garg K. Morphology
and immunohistochemistry for 2SC and FH aid in the detection of fumarate hydratase
gene aberrations in uterine leiomyomas from young patients. The American journal of
surgical pathology. 2015 1; 39(11):1529-39.
1. Bardella C, El‐Bahrawy M, Frizzell N, Adam J, Ternette N, Hatipoglu E, Howarth K,
O'flaherty L, Roberts I, Turner G, Taylor J. Aberrant succination of proteins in
fumarate hydratase‐deficient mice and HLRCC patients is a robust biomarker of
mutation status. The Journal of pathology. 2016; 225(1):4-11.
2. Baştuğ O, Kardaş F, Öztürk MA, Halis H, Memur Ş, Korkmaz L, Tağ Z, Güneş T. A
rare cause of opistotonus; fumaric aciduria: The first case presentation in Turkey.
Turkish Archives of Pediatrics/Türk Pediatri Arşivi. 2014; 49(1):74.
3. Bailey JP, Launonen V, Tomlinson IP. The FH mutation database: an online database
of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome
and congenital fumarase deficiency. BMC medical genetics. 2016; 9(1):20.
4. Briere JJ, Favier J, Gimenez-Roqueplo AP, Rustin P. Tricarboxylic acid cycle
dysfunction as a cause of human diseases and tumor formation. American Journal of
Physiology-cell physiology. 2016; 291(6): C1114-20.
5. Chan MM, Barnicoat A, Mumtaz F, Aitchison M, Side L, Brittain H, Bates AW, Gale
DP. Cascade Fumarate Hydratase mutation screening allows early detection of kidney
tumour: a case report. BMC medical genetics. 2017; 18(1):79.
6. Frezza C, Zheng L, Folger O, Rajagopalan KN, MacKenzie ED, Jerby L, Micaroni
M, Chaneton B, Adam J, Hedley A, Kalna G. Haem oxygenase is synthetically lethal
with the tumour suppressor fumarate hydratase. Nature. 2015; 477(7363):225.
7. Joseph NM, Solomon DA, Frizzell N, Rabban JT, Zaloudek C, Garg K. Morphology
and immunohistochemistry for 2SC and FH aid in the detection of fumarate hydratase
gene aberrations in uterine leiomyomas from young patients. The American journal of
surgical pathology. 2015 1; 39(11):1529-39.

Fumarase Deficiency 12
8. Kerrigan JF, Aleck KA, Tarby TJ, Bird CR, Heidenreich RA. Fumaric aciduria:
clinical and imaging features. Annals of neurology. 2013; 47(5):583-8.
9. Linehan WM, Rouault TA. Molecular pathways: fumarate hydratase-deficient kidney
cancer: targeting the Warburg effect in cancer. Clinical Cancer Research. 2013 30:
clincanres-0304.
10. Metternick-Jones SC, Lister KJ, Dawkins HJ, White CA, Weeramanthri TS. Review
of current international decision-making processes for newborn screening: lessons for
Australia. Frontiers in public health. 2015 10(3):214.
11. Mroch AR, Laudenschlager M, Flanagan JD. Detection of a novel FH whole gene
deletion in the propositus leading to subsequent prenatal diagnosis in a sibship with
fumarase deficiency. American Journal of Medical Genetics Part A. 2015;
158(1):155-8.
12. Ottolenghi C, Hubert L, Allanore Y, Brassier A, Altuzarra C, Mellot‐Draznieks C,
Bekri S, Goldenberg A, Veyrieres S, Boddaert N, Barbier V. Clinical and
biochemical heterogeneity associated with fumarase deficiency. Human mutation.
2014; 32(9):1046-52.
13. Picaud S, Kavanagh KL, Yue WW, Lee WH, Muller-Knapp S, Gileadi O, Sacchettini
J, Oppermann U. Structural basis of fumarate hydratase deficiency. Journal of
inherited metabolic disease. 2013 1; 34(3):671-6.
14. Sudarshan S, Sourbier C, Kong HS, Block K, Romero VA, Yang Y, Galindo C,
Mollapour M, Scroggins B, Goode N, Lee MJ. Fumarate hydratase deficiency in renal
cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1α
8. Kerrigan JF, Aleck KA, Tarby TJ, Bird CR, Heidenreich RA. Fumaric aciduria:
clinical and imaging features. Annals of neurology. 2013; 47(5):583-8.
9. Linehan WM, Rouault TA. Molecular pathways: fumarate hydratase-deficient kidney
cancer: targeting the Warburg effect in cancer. Clinical Cancer Research. 2013 30:
clincanres-0304.
10. Metternick-Jones SC, Lister KJ, Dawkins HJ, White CA, Weeramanthri TS. Review
of current international decision-making processes for newborn screening: lessons for
Australia. Frontiers in public health. 2015 10(3):214.
11. Mroch AR, Laudenschlager M, Flanagan JD. Detection of a novel FH whole gene
deletion in the propositus leading to subsequent prenatal diagnosis in a sibship with
fumarase deficiency. American Journal of Medical Genetics Part A. 2015;
158(1):155-8.
12. Ottolenghi C, Hubert L, Allanore Y, Brassier A, Altuzarra C, Mellot‐Draznieks C,
Bekri S, Goldenberg A, Veyrieres S, Boddaert N, Barbier V. Clinical and
biochemical heterogeneity associated with fumarase deficiency. Human mutation.
2014; 32(9):1046-52.
13. Picaud S, Kavanagh KL, Yue WW, Lee WH, Muller-Knapp S, Gileadi O, Sacchettini
J, Oppermann U. Structural basis of fumarate hydratase deficiency. Journal of
inherited metabolic disease. 2013 1; 34(3):671-6.
14. Sudarshan S, Sourbier C, Kong HS, Block K, Romero VA, Yang Y, Galindo C,
Mollapour M, Scroggins B, Goode N, Lee MJ. Fumarate hydratase deficiency in renal
cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1α
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide
1 out of 13



Your All-in-One AI-Powered Toolkit for Academic Success.
+13062052269
info@desklib.com
Available 24*7 on WhatsApp / Email
![[object Object]](/_next/static/media/star-bottom.7253800d.svg)
Unlock your academic potential
Copyright © 2020–2026 A2Z Services. All Rights Reserved. Developed and managed by ZUCOL.