In Silico Modeling Toxicology and SMILES Software Analysis
VerifiedAdded on 2022/09/01
|25
|7727
|74
Report
AI Summary
This report delves into the application of in silico modeling in toxicology, emphasizing its significance in reducing drug attrition rates during pharmaceutical research and development. It highlights the limitations of traditional methods and the advantages of in silico techniques, particularly the use of Quantitative Structure-Activity Relationship (QSAR) techniques and the SMILES (Simplified Molecular Input Line Entry System) software. The report discusses how in silico methods can identify potential toxicological issues early in the drug development process, thereby minimizing costly failures in later stages. The study showcases the role of in silico methods in addressing the high attrition rates, reducing animal testing, and improving the efficiency of drug discovery by predicting the toxicity of new drug compounds. The report further explores the ability of in silico methods to analyze drug safety, identify knowledge gaps, and provide computational solutions to address the weaknesses of drugs. The report also examines the role of in silico pharmacology and the importance of time and gain in the drug discovery process, making in silico methods a promising candidate in minimizing problems associated with late discovery. The report concludes by highlighting the need for discovery of shortcuts and rules to expedite the process of drug discovery, emphasizing the advantages of computational methods in providing real-time analysis of drugs during synthesis.

IN SILICO MODELLING TOXICOLOGY 1
IN SILICO MODELLING TOXICOLOGY
By [Name]
Course
Professor’s Name
Institution
Location of Institution
Date
IN SILICO MODELLING TOXICOLOGY
By [Name]
Course
Professor’s Name
Institution
Location of Institution
Date
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

IN SILICO MODELLING TOXICOLOGY 2
Introduction
The discovery of various drugs more often than note requires a significant investment in terms of
time and resources. It has been considered very significant to address the high attrition rates in
drug development. The pharmaceutical industries, therefore, remain under huge pressure to
provide solutions to the high rates of attrition in drug development.
It is imperative to note that, late-stage attrition drug candidates remain a problem. This is despite
the recent developments that have significantly reduced the number of a drug failing at the late
stage of development due to poor pharmacokinetic and toxickinetic profiles. Moreover, for drug
development to meet both financial and economic sustainability, there is a great need to reduce
animal testing for toxicological risk assessment. In silico methods, therefore, offer an alternative
that can be used to address the challenges associated with the high attrition rates in drug
development.
The modeling of various physiological properties of drugs such as toxicity, metabolism, drug-
drug interactions, and carcinogenesis have been facilitated by the use of Quantitative Structure-
Activity Relationship (QSAR) techniques. The application and use of QSAR in modeling various
physiological properties of drugs require the application of SMILES (Simplified Molecular Input
Line Entry System). SMILES is a scientific language usually developed to process information
on modern chemistry (Rognan, 2017, p.48). Moreover, this technique applies the principles of
molecular graph theory and very small and natural grammar to allow rigorous structure
specification.
Most of the highly efficient chemical computer applications are designed to generate unique
chemical notation to allow constant-speed database retrieval, flexible substructure searching, and
property prediction models through their ease of usage by the chemist and machine
Introduction
The discovery of various drugs more often than note requires a significant investment in terms of
time and resources. It has been considered very significant to address the high attrition rates in
drug development. The pharmaceutical industries, therefore, remain under huge pressure to
provide solutions to the high rates of attrition in drug development.
It is imperative to note that, late-stage attrition drug candidates remain a problem. This is despite
the recent developments that have significantly reduced the number of a drug failing at the late
stage of development due to poor pharmacokinetic and toxickinetic profiles. Moreover, for drug
development to meet both financial and economic sustainability, there is a great need to reduce
animal testing for toxicological risk assessment. In silico methods, therefore, offer an alternative
that can be used to address the challenges associated with the high attrition rates in drug
development.
The modeling of various physiological properties of drugs such as toxicity, metabolism, drug-
drug interactions, and carcinogenesis have been facilitated by the use of Quantitative Structure-
Activity Relationship (QSAR) techniques. The application and use of QSAR in modeling various
physiological properties of drugs require the application of SMILES (Simplified Molecular Input
Line Entry System). SMILES is a scientific language usually developed to process information
on modern chemistry (Rognan, 2017, p.48). Moreover, this technique applies the principles of
molecular graph theory and very small and natural grammar to allow rigorous structure
specification.
Most of the highly efficient chemical computer applications are designed to generate unique
chemical notation to allow constant-speed database retrieval, flexible substructure searching, and
property prediction models through their ease of usage by the chemist and machine

IN SILICO MODELLING TOXICOLOGY 3
compatibility. The use and application of SMILES in predicting molecular structure are due to
several advantages. Due to its ease of use and accessibility by chemists, interpretation and
generation of chemical notation that are independent of the specific computer system in use is
significantly simplified (Naven, & Louise-May, 2015, p.1306). The ability of SMILES to
represent molecular structure using a linear string of symbols similar to natural language makes
is use very popular by most computational chemist in predicting molecular structure.
SMILES software simplifies the chemical structure of chemical compounds through a two-
dimensional valence-oriented picture graph. It is worth noting that, SMILES notation is a group
of characters that end with space used in the prediction of molecular structure. Hydrogen can
either be included or excluded in the representation of molecular structure using this software
(Worth, 2018). The use of SMILES in predicting the chemical structure of compounds follows
certain rules and specifications. Letters are always used to represent the atoms within the
structure as shown below.
C Ethane (C2H6)
N ammine (NH2)
Single, double, triple, and aromatic bonds are usually represented in the SMILES software using
-, =, #, respectively. Branches, on the other hand, are specified by closures in parenthesis. See the
example below.
SMILES notation has been considered by most researchers as the most favourable software for
developing a large database as well as elucidating molecular structures (Naven, & Louise-May,
2015, p.1306). In sum, the development of molecular descriptors that are directly calculated from
compatibility. The use and application of SMILES in predicting molecular structure are due to
several advantages. Due to its ease of use and accessibility by chemists, interpretation and
generation of chemical notation that are independent of the specific computer system in use is
significantly simplified (Naven, & Louise-May, 2015, p.1306). The ability of SMILES to
represent molecular structure using a linear string of symbols similar to natural language makes
is use very popular by most computational chemist in predicting molecular structure.
SMILES software simplifies the chemical structure of chemical compounds through a two-
dimensional valence-oriented picture graph. It is worth noting that, SMILES notation is a group
of characters that end with space used in the prediction of molecular structure. Hydrogen can
either be included or excluded in the representation of molecular structure using this software
(Worth, 2018). The use of SMILES in predicting the chemical structure of compounds follows
certain rules and specifications. Letters are always used to represent the atoms within the
structure as shown below.
C Ethane (C2H6)
N ammine (NH2)
Single, double, triple, and aromatic bonds are usually represented in the SMILES software using
-, =, #, respectively. Branches, on the other hand, are specified by closures in parenthesis. See the
example below.
SMILES notation has been considered by most researchers as the most favourable software for
developing a large database as well as elucidating molecular structures (Naven, & Louise-May,
2015, p.1306). In sum, the development of molecular descriptors that are directly calculated from
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

IN SILICO MODELLING TOXICOLOGY 4
the SMILES for use in predictive toxicology, therefore, makes SMILES notation to be an
attractive approach in the QSAR/QSPR modeling research. As earlier mentioned, there is a great
need to provide solutions to the escalating rates of drug failing in both early and late stages of
drug development as well as the need to reduce animal testing for toxicological risk assessment.
The study, therefore, investigates the essential role played by in silico modeling toxicology
technique in conjunction with SMILES software to reduce drug attrition as justified below.
Role of in silico technique in reducing drug attrition.
The rate at which new drugs are failing in pharmaceutical research and development has been
significantly reduced due to the application of the in silico predictive toxicology. This is due to
its ability to identify the gaps that exist in the current mechanistic and chemical toxicology used
in the testing of drug toxicity levels (Atienzar et al 2016). Computational toxicologist has,
therefore, engaged in understanding these gaps to provide the solution to factors that lead to high
drug attrition rate.
The efficiency of costly drug discovery can be improved by predicting the toxicity of any new
drug compound. Many predictive tools that were previously used in predicting the toxicity of
drugs have been withdrawn from the market due to safety reasons. The UK markets recently
withdraw nefazodone from the market owing to severe, albeit rare, hepatotoxicity level (Atienzar
et al 2016). The several safety liabilities associated with nefazodone, therefore, led to its failure
in the late stages of development. Moreover, chloroanaline moiety is also currently undergoing
extensive metabolism to form reactive intermediates due to the already identified structural alerts
in the drug.
the SMILES for use in predictive toxicology, therefore, makes SMILES notation to be an
attractive approach in the QSAR/QSPR modeling research. As earlier mentioned, there is a great
need to provide solutions to the escalating rates of drug failing in both early and late stages of
drug development as well as the need to reduce animal testing for toxicological risk assessment.
The study, therefore, investigates the essential role played by in silico modeling toxicology
technique in conjunction with SMILES software to reduce drug attrition as justified below.
Role of in silico technique in reducing drug attrition.
The rate at which new drugs are failing in pharmaceutical research and development has been
significantly reduced due to the application of the in silico predictive toxicology. This is due to
its ability to identify the gaps that exist in the current mechanistic and chemical toxicology used
in the testing of drug toxicity levels (Atienzar et al 2016). Computational toxicologist has,
therefore, engaged in understanding these gaps to provide the solution to factors that lead to high
drug attrition rate.
The efficiency of costly drug discovery can be improved by predicting the toxicity of any new
drug compound. Many predictive tools that were previously used in predicting the toxicity of
drugs have been withdrawn from the market due to safety reasons. The UK markets recently
withdraw nefazodone from the market owing to severe, albeit rare, hepatotoxicity level (Atienzar
et al 2016). The several safety liabilities associated with nefazodone, therefore, led to its failure
in the late stages of development. Moreover, chloroanaline moiety is also currently undergoing
extensive metabolism to form reactive intermediates due to the already identified structural alerts
in the drug.
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

IN SILICO MODELLING TOXICOLOGY 5
Figure1: showing the metabolic oxidation of nefazodone to form a quinone-imide as a reactive
intermediate (Naven, & Louise-May, 2015, p.1304).
The serious organ toxicities caused by the chloroanaline moiety, therefore, calls for the quick
implementation of in silico tools to address the high level of organ toxicity caused by these drugs
(Rezaee and Abdollahi, 2017, p.104). Moreover, the inability of the in Vitro system to
effectively assess drug safety before released into the market calls for the use and application of
in silico in the testing of toxicity in drugs.
The high level of off-target toxicity has further led to the failure of other drugs such as
ximelgatran, an antithrombotic and anticoagulant. Such drugs were recently removed from the
UK markets owing to their high liver toxicity level (Richardson et al., 2017, p. 544). The failure
of these drugs and final removal from the market is due to a lack of signals on their toxicity
liability during the preclinical or clinical screening as a result of lack of assays and clarity on the
underlying mechanisms at the time of their development (Naven, & Louise-May, 2015, p.1306).
The failure of such drugs, therefore, significantly triggered the implantation of in silico methods
to reduce drug attrition especially during their late stages of development.
In silico models tend to provide solutions to drug failure by identifying the gaps that exist in the
current safety screening cascades through understanding the structural, mechanistic, chemical,
and the pharmacological space of the drug molecules.
The model contributes significantly in minimizing the rate of drug failure by identifying the
toxicological knowledge gaps that exist in the already exposed miss-predicted compounds
Figure1: showing the metabolic oxidation of nefazodone to form a quinone-imide as a reactive
intermediate (Naven, & Louise-May, 2015, p.1304).
The serious organ toxicities caused by the chloroanaline moiety, therefore, calls for the quick
implementation of in silico tools to address the high level of organ toxicity caused by these drugs
(Rezaee and Abdollahi, 2017, p.104). Moreover, the inability of the in Vitro system to
effectively assess drug safety before released into the market calls for the use and application of
in silico in the testing of toxicity in drugs.
The high level of off-target toxicity has further led to the failure of other drugs such as
ximelgatran, an antithrombotic and anticoagulant. Such drugs were recently removed from the
UK markets owing to their high liver toxicity level (Richardson et al., 2017, p. 544). The failure
of these drugs and final removal from the market is due to a lack of signals on their toxicity
liability during the preclinical or clinical screening as a result of lack of assays and clarity on the
underlying mechanisms at the time of their development (Naven, & Louise-May, 2015, p.1306).
The failure of such drugs, therefore, significantly triggered the implantation of in silico methods
to reduce drug attrition especially during their late stages of development.
In silico models tend to provide solutions to drug failure by identifying the gaps that exist in the
current safety screening cascades through understanding the structural, mechanistic, chemical,
and the pharmacological space of the drug molecules.
The model contributes significantly in minimizing the rate of drug failure by identifying the
toxicological knowledge gaps that exist in the already exposed miss-predicted compounds
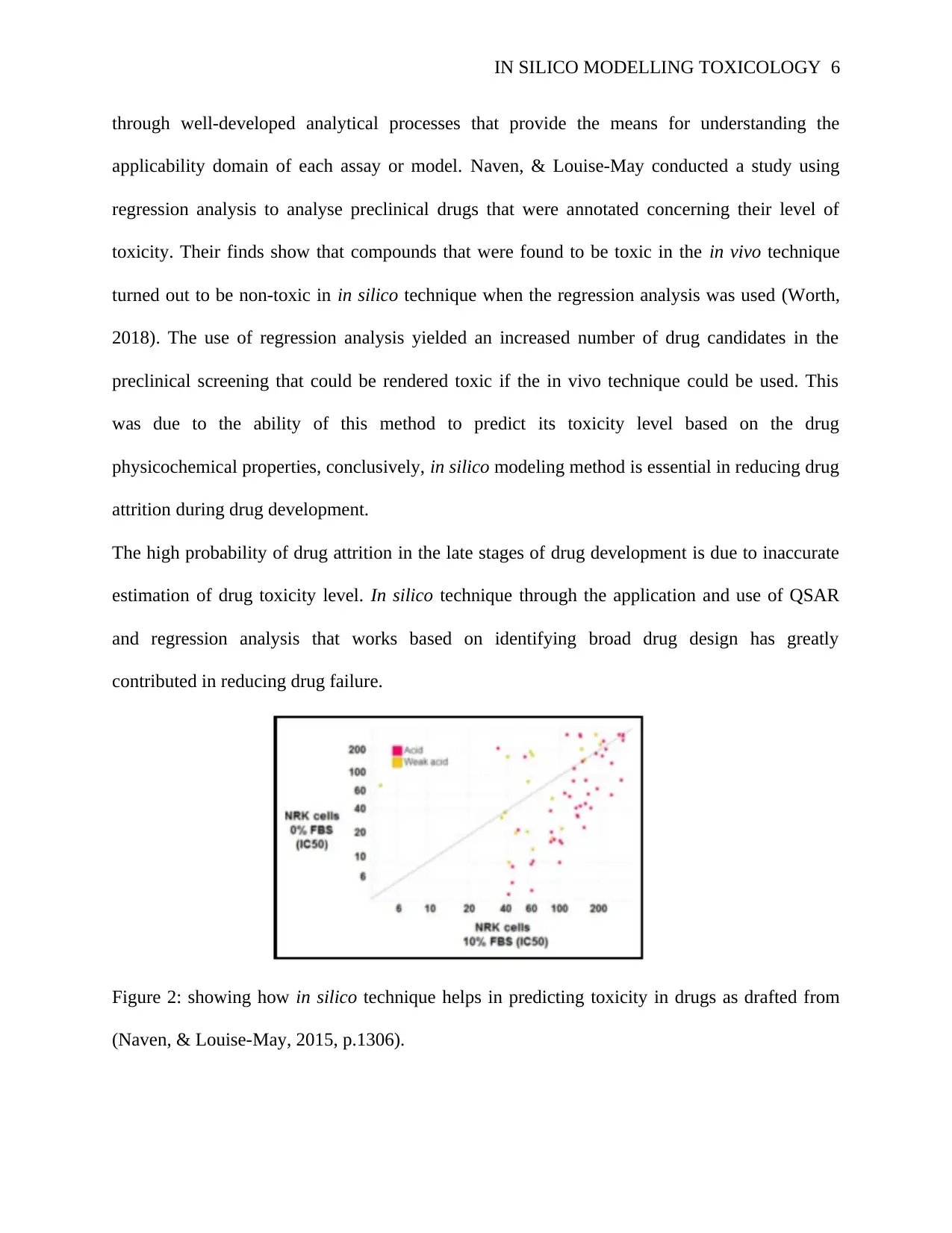
IN SILICO MODELLING TOXICOLOGY 6
through well-developed analytical processes that provide the means for understanding the
applicability domain of each assay or model. Naven, & Louise-May conducted a study using
regression analysis to analyse preclinical drugs that were annotated concerning their level of
toxicity. Their finds show that compounds that were found to be toxic in the in vivo technique
turned out to be non-toxic in in silico technique when the regression analysis was used (Worth,
2018). The use of regression analysis yielded an increased number of drug candidates in the
preclinical screening that could be rendered toxic if the in vivo technique could be used. This
was due to the ability of this method to predict its toxicity level based on the drug
physicochemical properties, conclusively, in silico modeling method is essential in reducing drug
attrition during drug development.
The high probability of drug attrition in the late stages of drug development is due to inaccurate
estimation of drug toxicity level. In silico technique through the application and use of QSAR
and regression analysis that works based on identifying broad drug design has greatly
contributed in reducing drug failure.
Figure 2: showing how in silico technique helps in predicting toxicity in drugs as drafted from
(Naven, & Louise-May, 2015, p.1306).
through well-developed analytical processes that provide the means for understanding the
applicability domain of each assay or model. Naven, & Louise-May conducted a study using
regression analysis to analyse preclinical drugs that were annotated concerning their level of
toxicity. Their finds show that compounds that were found to be toxic in the in vivo technique
turned out to be non-toxic in in silico technique when the regression analysis was used (Worth,
2018). The use of regression analysis yielded an increased number of drug candidates in the
preclinical screening that could be rendered toxic if the in vivo technique could be used. This
was due to the ability of this method to predict its toxicity level based on the drug
physicochemical properties, conclusively, in silico modeling method is essential in reducing drug
attrition during drug development.
The high probability of drug attrition in the late stages of drug development is due to inaccurate
estimation of drug toxicity level. In silico technique through the application and use of QSAR
and regression analysis that works based on identifying broad drug design has greatly
contributed in reducing drug failure.
Figure 2: showing how in silico technique helps in predicting toxicity in drugs as drafted from
(Naven, & Louise-May, 2015, p.1306).
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

IN SILICO MODELLING TOXICOLOGY 7
Different categories of assays that describe the safety liabilities of acidic drugs can be effectively
identified using the above regression analysis. Based on the results from the cytotoxicity, in
silico method is a chief player in determining the level of toxicity in acidic drugs.
The above figure shows the results obtained from several acidic drugs screened in a cytotoxicity
assay using NPK52E cells. The apparent cytotoxicity of acidic compounds is enhanced by the
reduced level of fetal bovine serum from the downward shift (Richardson et al., 2017, p. 544).
The result obtained from the above regression analysis help in identification of compounds that
cause significant in silico toxicity hence reducing drug attrition rate.
The ability to avoid future attrition in drug discovery significantly depends on addressing the
knowledge gap identified by the in silico methods. In silico methods address these toxicological
knowledge gaps identified through provision of computational solutions to the weaknesses
observed in the drugs withdrawn from the markets for safety reasons.
In silico can be defined as the process of integration of modern computing and information
technology to improve the process of assessing drugs. (Baur et al., 2020, p.28). It uses computer
or computer simulation models to perform experimentation to test drug toxicology. In silico
differ significantly with other traditional toxicology methods. This is due the large number of
chemicals that are studied, breadth of endpoints and pathways covered, levels of biological
organization examined and the range of exposure conditions considered during toxicity.
In silico pharmacology is increasingly becoming relevant in the field of drug discovery due to its
ability to use software such as SMILES in capturing, analysing, and integrating biological and
medical data from different sources (Prior et al, 2019, p. 12). The method plays a major role in
the reduction of drug attrition during drug development. Toxicity can be defined as the
processing of measuring the side effects of drugs before released into the clinics and further into
Different categories of assays that describe the safety liabilities of acidic drugs can be effectively
identified using the above regression analysis. Based on the results from the cytotoxicity, in
silico method is a chief player in determining the level of toxicity in acidic drugs.
The above figure shows the results obtained from several acidic drugs screened in a cytotoxicity
assay using NPK52E cells. The apparent cytotoxicity of acidic compounds is enhanced by the
reduced level of fetal bovine serum from the downward shift (Richardson et al., 2017, p. 544).
The result obtained from the above regression analysis help in identification of compounds that
cause significant in silico toxicity hence reducing drug attrition rate.
The ability to avoid future attrition in drug discovery significantly depends on addressing the
knowledge gap identified by the in silico methods. In silico methods address these toxicological
knowledge gaps identified through provision of computational solutions to the weaknesses
observed in the drugs withdrawn from the markets for safety reasons.
In silico can be defined as the process of integration of modern computing and information
technology to improve the process of assessing drugs. (Baur et al., 2020, p.28). It uses computer
or computer simulation models to perform experimentation to test drug toxicology. In silico
differ significantly with other traditional toxicology methods. This is due the large number of
chemicals that are studied, breadth of endpoints and pathways covered, levels of biological
organization examined and the range of exposure conditions considered during toxicity.
In silico pharmacology is increasingly becoming relevant in the field of drug discovery due to its
ability to use software such as SMILES in capturing, analysing, and integrating biological and
medical data from different sources (Prior et al, 2019, p. 12). The method plays a major role in
the reduction of drug attrition during drug development. Toxicity can be defined as the
processing of measuring the side effects of drugs before released into the clinics and further into
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

IN SILICO MODELLING TOXICOLOGY 8
the market. It can also be defined as the level of potential effects drugs or chemicals cause on
their final users either through single-exposure or multiple-exposure (Troth et al., 2019, p. 128).
Toxicity endpoints are the specific categories of side effects caused by drug testing such as both
qualitative and quantitative carcinogenicity. Toxicity tests, on the other hand, are the
identification of harmful effects caused by substances on plants, animals, and human beings.
In vitro toxicity tests is very advanced in the application and use of high throughput screening.
This characteristic makes it to heavily really on the use of animals for toxicity testing. In silico
toxicology, on the other hand, rely on computerized techniques such as methods, algorithm,
software, and data for toxicity assessment (Patel et al, 2019, p.8). Toxicity testing in silico
toxicology involves drug organization, analysis, modeling, simulation, and prediction of its
structure.
In silico methods analyses the side effects of any drug by intertwining the drug or chemical
within component of the in silico pharmacology. This is aided by the use of databases obtained
from the computer (Naven, & Louise-May, 2015, p.1306). It makes predictions on the suggested
hypotheses, and ultimately provide discoveries in the field of drug discoveries by creating
computational models or simulations.
Time and gain significantly play a critical role in any successful industry. Drug discovery firms
are such firms with huge and complex information to handle and interpret and therefore need to
save on time for again in the discovery process. For the newly discovered drugs to proceed
quickly into the clinic and the market, there is a great need for discovery of shortcuts and rules
(Parthasarathi, and Dhawan, 2018, p.91). In silico method is a promising candidate in ensuring
problems associated with late discovery are minimized.
the market. It can also be defined as the level of potential effects drugs or chemicals cause on
their final users either through single-exposure or multiple-exposure (Troth et al., 2019, p. 128).
Toxicity endpoints are the specific categories of side effects caused by drug testing such as both
qualitative and quantitative carcinogenicity. Toxicity tests, on the other hand, are the
identification of harmful effects caused by substances on plants, animals, and human beings.
In vitro toxicity tests is very advanced in the application and use of high throughput screening.
This characteristic makes it to heavily really on the use of animals for toxicity testing. In silico
toxicology, on the other hand, rely on computerized techniques such as methods, algorithm,
software, and data for toxicity assessment (Patel et al, 2019, p.8). Toxicity testing in silico
toxicology involves drug organization, analysis, modeling, simulation, and prediction of its
structure.
In silico methods analyses the side effects of any drug by intertwining the drug or chemical
within component of the in silico pharmacology. This is aided by the use of databases obtained
from the computer (Naven, & Louise-May, 2015, p.1306). It makes predictions on the suggested
hypotheses, and ultimately provide discoveries in the field of drug discoveries by creating
computational models or simulations.
Time and gain significantly play a critical role in any successful industry. Drug discovery firms
are such firms with huge and complex information to handle and interpret and therefore need to
save on time for again in the discovery process. For the newly discovered drugs to proceed
quickly into the clinic and the market, there is a great need for discovery of shortcuts and rules
(Parthasarathi, and Dhawan, 2018, p.91). In silico method is a promising candidate in ensuring
problems associated with late discovery are minimized.

IN SILICO MODELLING TOXICOLOGY 9
The high rate at which the discovered drugs fail to reach the clinic and into the market is as a
result of the high cost involved and a lot of time consumed in the process assessing the risks
associated with such drugs. This, therefore, calls for the use and application of in silico
computational method. Computational methods has the ability to provide real time analysis of
drugs during their synthesis. This again gives it a unique advantage over in vitro and in vivo. The
application of this computational method, therefore, is very significant in terms of
complimenting other methods used in the certification process of drugs.
In silico toxicology employs several computational tools in the reduction of attrition in a drug.
They include, toxicity database of compounds, various simulation tools, and modeling tools such
as statistical packages (Lucas et al., 2019, p.1313).
Figure 1: Showing all the steps involved in the generation of prediction models (Kenna, and
Ram, 2019, 168).
In silico toxicology also employs several methods or models in its role in reducing drug attrition.
They include, pharmacokinetic models, pharmacodynamics model, and Quantitative Structure-
Activity Relationship, and Quantity Structure Toxicity/Property Relationship (Przekwas and
The high rate at which the discovered drugs fail to reach the clinic and into the market is as a
result of the high cost involved and a lot of time consumed in the process assessing the risks
associated with such drugs. This, therefore, calls for the use and application of in silico
computational method. Computational methods has the ability to provide real time analysis of
drugs during their synthesis. This again gives it a unique advantage over in vitro and in vivo. The
application of this computational method, therefore, is very significant in terms of
complimenting other methods used in the certification process of drugs.
In silico toxicology employs several computational tools in the reduction of attrition in a drug.
They include, toxicity database of compounds, various simulation tools, and modeling tools such
as statistical packages (Lucas et al., 2019, p.1313).
Figure 1: Showing all the steps involved in the generation of prediction models (Kenna, and
Ram, 2019, 168).
In silico toxicology also employs several methods or models in its role in reducing drug attrition.
They include, pharmacokinetic models, pharmacodynamics model, and Quantitative Structure-
Activity Relationship, and Quantity Structure Toxicity/Property Relationship (Przekwas and
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

IN SILICO MODELLING TOXICOLOGY 10
Somayaji, 2018, p.311). The study heavily relies on QASR/QSPR models in illustrating the role
played by in silico toxicology in reducing drug attrition.
There are two main broad categories of in silico testing methods. They include, the expert
system and the predictive systems. Global or expert systems predict toxicity in compounds by
mimicking human reasoning and formalizing the existing knowledge while predictive expert
systems predict toxicity in compounds through the application of various models (Richardson et
al., 2017, p. 544). Quantitative Structure-Activity Relationship (QSAR) is an example of such
models that predicts chemicals’ toxicity through the use of molecular descriptors. It can be
defined as the process of correlating a quantitative measure of chemical structure to either a
physical property or biological effects such as toxic outcomes using a mathematical model.
QSAR makes predictions about the toxicity level of a substance using various parameters also
known as molecular descriptors. Molecular descriptors are the numerical representation of the
molecular structure. Apart from descriptors, QSAR also employs the use of linear regression
analysis as well as other multivariate statistical techniques to make the prediction (Loiodice,
Nogueira da Costa, and Atienzar, 2019, p.113). Moreover, QSAR applies the use of molecular
descriptors to make prediction on the level of toxicity during drug development by calculating
the ligands and their target macromolecules atomic interactions. These atomic interactions forms
the basis through which the level of toxicity in drugs is determined by the QSAR.
The use of the QSAR model to make predictions in in silico methods involves grouping chemical
and filing of data gaps. The grouping of these drugs takes into account their rate of reaction and
structural similarity (Ivanov et al., 2016, p.58). QSAR also relies on the differences in the
chemical reactivity of molecules to make its prediction in the assumption that, the analog and
target molecules show toxicology significant metabolic by either converging or diverging.
Somayaji, 2018, p.311). The study heavily relies on QASR/QSPR models in illustrating the role
played by in silico toxicology in reducing drug attrition.
There are two main broad categories of in silico testing methods. They include, the expert
system and the predictive systems. Global or expert systems predict toxicity in compounds by
mimicking human reasoning and formalizing the existing knowledge while predictive expert
systems predict toxicity in compounds through the application of various models (Richardson et
al., 2017, p. 544). Quantitative Structure-Activity Relationship (QSAR) is an example of such
models that predicts chemicals’ toxicity through the use of molecular descriptors. It can be
defined as the process of correlating a quantitative measure of chemical structure to either a
physical property or biological effects such as toxic outcomes using a mathematical model.
QSAR makes predictions about the toxicity level of a substance using various parameters also
known as molecular descriptors. Molecular descriptors are the numerical representation of the
molecular structure. Apart from descriptors, QSAR also employs the use of linear regression
analysis as well as other multivariate statistical techniques to make the prediction (Loiodice,
Nogueira da Costa, and Atienzar, 2019, p.113). Moreover, QSAR applies the use of molecular
descriptors to make prediction on the level of toxicity during drug development by calculating
the ligands and their target macromolecules atomic interactions. These atomic interactions forms
the basis through which the level of toxicity in drugs is determined by the QSAR.
The use of the QSAR model to make predictions in in silico methods involves grouping chemical
and filing of data gaps. The grouping of these drugs takes into account their rate of reaction and
structural similarity (Ivanov et al., 2016, p.58). QSAR also relies on the differences in the
chemical reactivity of molecules to make its prediction in the assumption that, the analog and
target molecules show toxicology significant metabolic by either converging or diverging.
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

IN SILICO MODELLING TOXICOLOGY 11
Moreover, it considers the effects on the bioavailability and consequently biological responses
by identifying the differences in the physicochemical properties. In sum, QSAR works by
applying forming chemical categories and then using the measured data on a few categories to
estimate the missing values for the untested member.
It’s always believed that chemicals that can work through the QSAR mechanism can also fit its
model. Future vector of chemicals properties (θP) and a function f can be applied to predict
toxicity given by the formula
T = f (θP)
It is imperative to note that, the generation of a local QSAR is usually from congeneric
chemicals. Diverse chemicals, on the other hand, are used to generate a global QSAR model.
Since the local QSAR is customized for specific chemicals, they are more accurate compared to
the global QSAR. Global QSAR model tends to be more practical but less accurate, this is
because there is an overhead in developing the local ones for each type of chemical (Hodos et al,
2016, p.186). Quantity Structure Toxicity/Property Relationship are types of QSAR models used
for predicting toxicity and chemical properties respectively.
Developing of a QSAR model involves two main steps. The molecular descriptors are first
generated followed by the generation of the models to fit the data. The selection of algorithms
can be achieved through the use of simulated annealing, genetic algorithm, or principal
component analysis (Clippinger et al., 2018, p.53). Two dimensional scatter plots of each
descriptor versus the biological activity can be used for the identification of significant
descriptors when there are a small number of descriptors.
Moreover, it considers the effects on the bioavailability and consequently biological responses
by identifying the differences in the physicochemical properties. In sum, QSAR works by
applying forming chemical categories and then using the measured data on a few categories to
estimate the missing values for the untested member.
It’s always believed that chemicals that can work through the QSAR mechanism can also fit its
model. Future vector of chemicals properties (θP) and a function f can be applied to predict
toxicity given by the formula
T = f (θP)
It is imperative to note that, the generation of a local QSAR is usually from congeneric
chemicals. Diverse chemicals, on the other hand, are used to generate a global QSAR model.
Since the local QSAR is customized for specific chemicals, they are more accurate compared to
the global QSAR. Global QSAR model tends to be more practical but less accurate, this is
because there is an overhead in developing the local ones for each type of chemical (Hodos et al,
2016, p.186). Quantity Structure Toxicity/Property Relationship are types of QSAR models used
for predicting toxicity and chemical properties respectively.
Developing of a QSAR model involves two main steps. The molecular descriptors are first
generated followed by the generation of the models to fit the data. The selection of algorithms
can be achieved through the use of simulated annealing, genetic algorithm, or principal
component analysis (Clippinger et al., 2018, p.53). Two dimensional scatter plots of each
descriptor versus the biological activity can be used for the identification of significant
descriptors when there are a small number of descriptors.
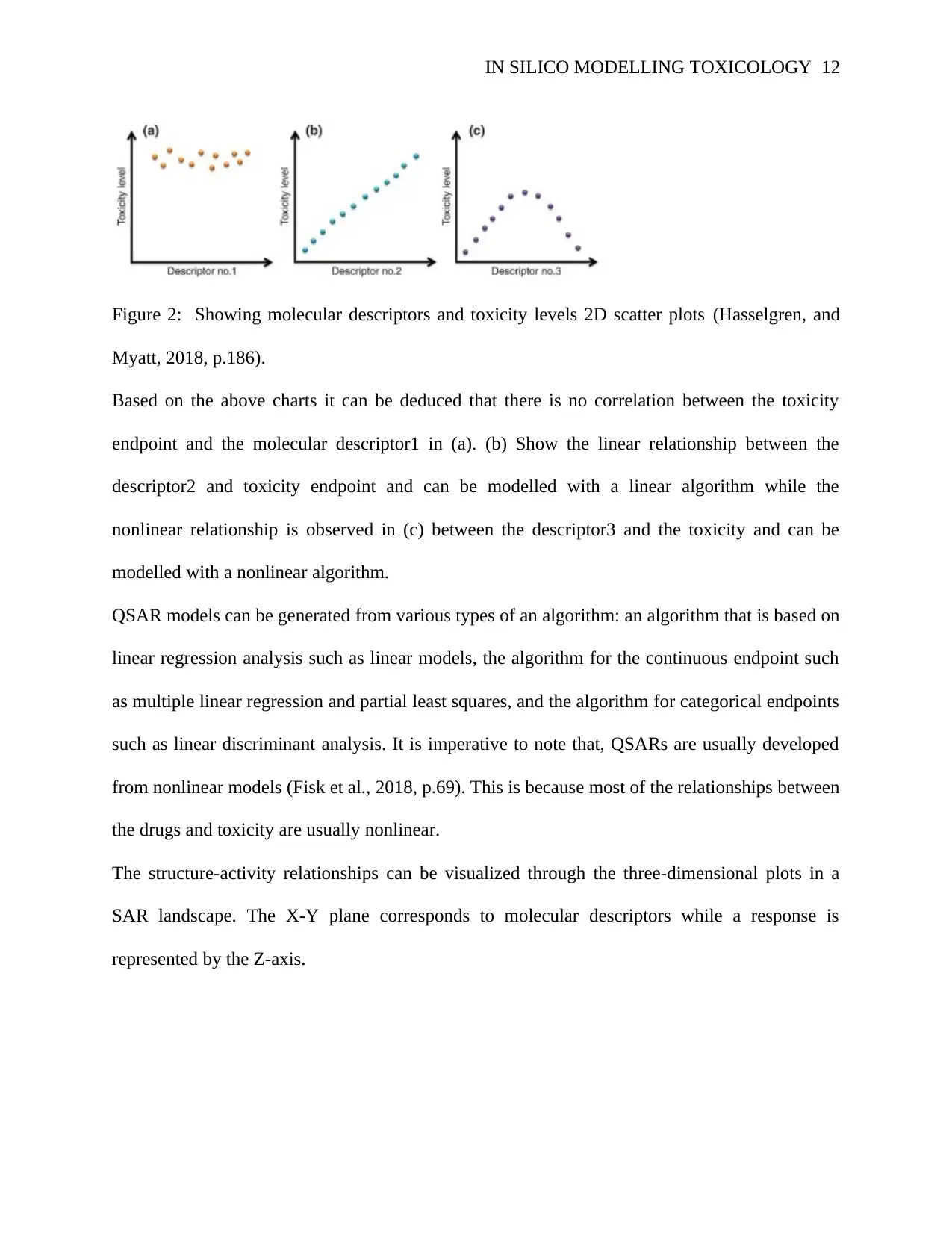
IN SILICO MODELLING TOXICOLOGY 12
Figure 2: Showing molecular descriptors and toxicity levels 2D scatter plots (Hasselgren, and
Myatt, 2018, p.186).
Based on the above charts it can be deduced that there is no correlation between the toxicity
endpoint and the molecular descriptor1 in (a). (b) Show the linear relationship between the
descriptor2 and toxicity endpoint and can be modelled with a linear algorithm while the
nonlinear relationship is observed in (c) between the descriptor3 and the toxicity and can be
modelled with a nonlinear algorithm.
QSAR models can be generated from various types of an algorithm: an algorithm that is based on
linear regression analysis such as linear models, the algorithm for the continuous endpoint such
as multiple linear regression and partial least squares, and the algorithm for categorical endpoints
such as linear discriminant analysis. It is imperative to note that, QSARs are usually developed
from nonlinear models (Fisk et al., 2018, p.69). This is because most of the relationships between
the drugs and toxicity are usually nonlinear.
The structure-activity relationships can be visualized through the three-dimensional plots in a
SAR landscape. The X-Y plane corresponds to molecular descriptors while a response is
represented by the Z-axis.
Figure 2: Showing molecular descriptors and toxicity levels 2D scatter plots (Hasselgren, and
Myatt, 2018, p.186).
Based on the above charts it can be deduced that there is no correlation between the toxicity
endpoint and the molecular descriptor1 in (a). (b) Show the linear relationship between the
descriptor2 and toxicity endpoint and can be modelled with a linear algorithm while the
nonlinear relationship is observed in (c) between the descriptor3 and the toxicity and can be
modelled with a nonlinear algorithm.
QSAR models can be generated from various types of an algorithm: an algorithm that is based on
linear regression analysis such as linear models, the algorithm for the continuous endpoint such
as multiple linear regression and partial least squares, and the algorithm for categorical endpoints
such as linear discriminant analysis. It is imperative to note that, QSARs are usually developed
from nonlinear models (Fisk et al., 2018, p.69). This is because most of the relationships between
the drugs and toxicity are usually nonlinear.
The structure-activity relationships can be visualized through the three-dimensional plots in a
SAR landscape. The X-Y plane corresponds to molecular descriptors while a response is
represented by the Z-axis.
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide
1 out of 25

Your All-in-One AI-Powered Toolkit for Academic Success.
+13062052269
info@desklib.com
Available 24*7 on WhatsApp / Email
![[object Object]](/_next/static/media/star-bottom.7253800d.svg)
Unlock your academic potential
Copyright © 2020–2025 A2Z Services. All Rights Reserved. Developed and managed by ZUCOL.