Deakin University HMM301: Pharmacology Report on Ivacaftor's Mechanism
VerifiedAdded on 2023/01/23
|7
|2392
|78
Report
AI Summary
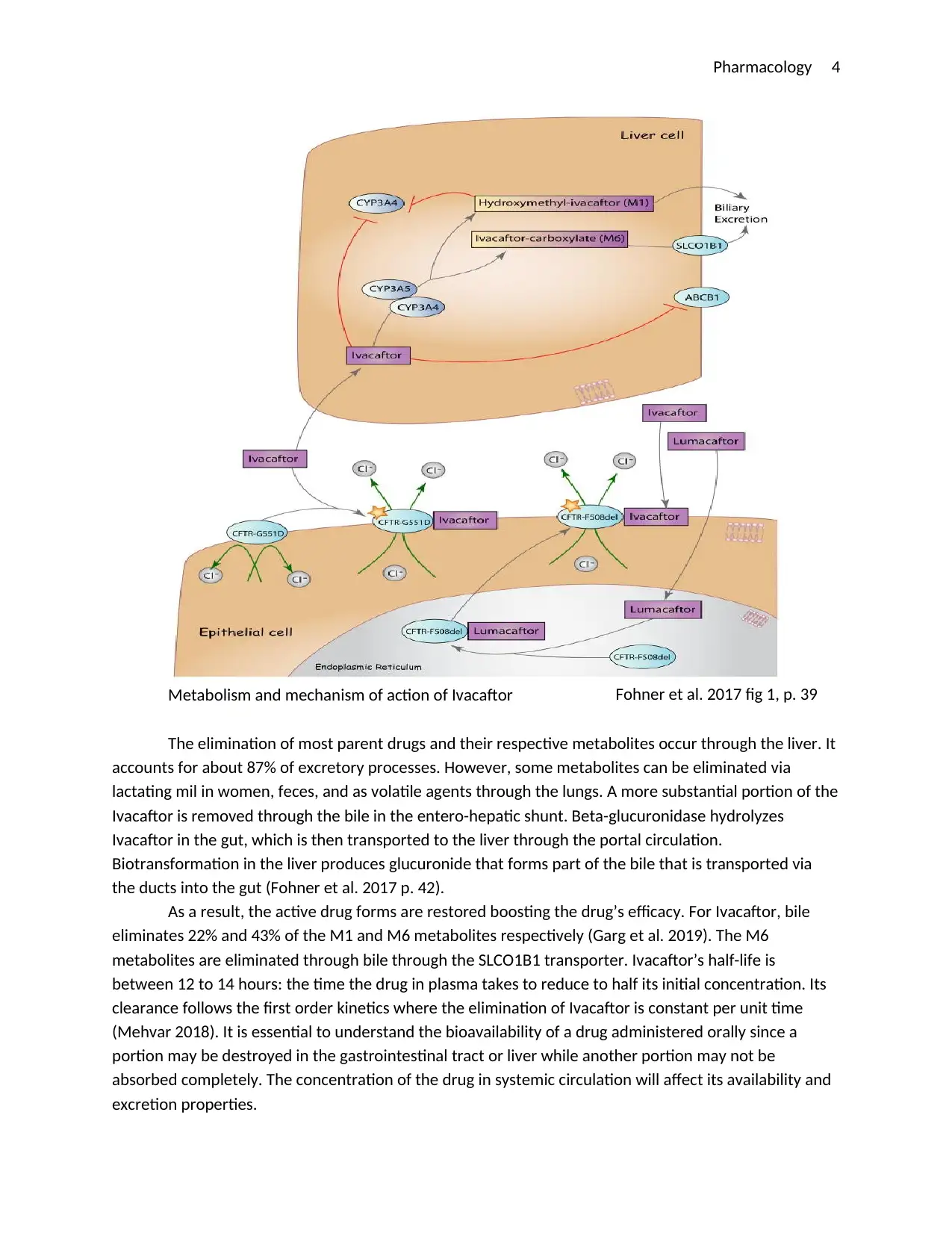
This report delves into the pharmacology of Ivacaftor, a CFTR potentiator used in the treatment of cystic fibrosis. It begins with an introduction to Ivacaftor, its mechanism of action, and its role in treating cystic fibrosis, a genetic disorder characterized by thick mucus in the lungs and digestive system. The report then explores the drug's pharmacodynamics, detailing its interaction with the CFTR protein and its impact on chloride ion transport, which is essential for restoring the function of epithelial cells in the lungs, pancreas, and other organs. The report also discusses the drug's pharmacokinetics, including its absorption, distribution, metabolism, and elimination processes. It highlights the importance of administering Ivacaftor with fatty foods to enhance its bioavailability and describes how the liver metabolizes the drug, producing both active and inactive metabolites. The report concludes with a summary of Ivacaftor's effectiveness in treating cystic fibrosis and its potential for future research. This report is based on the assignment brief for HMM301 Principles of Pharmacology at Deakin University.






1 out of 7
Related Documents






Your All-in-One AI-Powered Toolkit for Academic Success.
+13062052269
info@desklib.com
Available 24*7 on WhatsApp / Email
![[object Object]](/_next/static/media/star-bottom.7253800d.svg)
Copyright © 2020–2026 A2Z Services. All Rights Reserved. Developed and managed by ZUCOL.