The Pathogenesis of Insulin Resistance: Integrating Signaling Pathways and Substrate Flux
Added on 2023-05-29
12 Pages11989 Words158 Views
The pathogenesis of insulin resistance:
integrating signaling pathways and substrate
flux
Varman T. Samuel, Gerald I. Shulman
J Clin Invest. 2016;126(1):12-22. https://doi.org/10.1172/JCI77812.
Insulin resistance arises when the nutrient storage pathways evolved to maximize efficient
energy utilization are exposed to chronic energy surplus. Ectopic lipid accumulation in liver
and skeletal muscle triggers pathways that impair insulin signaling, leading to reduced
muscle glucose uptake and decreased hepatic glycogen synthesis. Muscle insulin
resistance, due to ectopic lipid, precedes liver insulin resistance and diverts ingested
glucose to the liver, resulting in increased hepatic de novo lipogenesis and hyperlipidemia.
Subsequent macrophage infiltration into white adipose tissue (WAT) leads to increased
lipolysis, which further increases hepatic triglyceride synthesis and hyperlipidemia due to
increased fatty acid esterification. Macrophage-induced WAT lipolysis also stimulates
hepatic gluconeogenesis, promoting fasting and postprandial hyperglycemia through
increased fatty acid delivery to the liver, which results in increased hepatic acetyl-CoA
content, a potent activator of pyruvate carboxylase, and increased glycerol conversion to
glucose. These substrate-regulated processes are mostly independent of insulin signaling
in the liver but are dependent on insulin signaling in WAT, which becomes defective with
inflammation. Therapies that decrease ectopic lipid storage and diminish macrophage-
induced WAT lipolysis will reverse the root causes of type 2 diabetes.
Review
Find the latest version:
http://jci.me/77812/pdf
integrating signaling pathways and substrate
flux
Varman T. Samuel, Gerald I. Shulman
J Clin Invest. 2016;126(1):12-22. https://doi.org/10.1172/JCI77812.
Insulin resistance arises when the nutrient storage pathways evolved to maximize efficient
energy utilization are exposed to chronic energy surplus. Ectopic lipid accumulation in liver
and skeletal muscle triggers pathways that impair insulin signaling, leading to reduced
muscle glucose uptake and decreased hepatic glycogen synthesis. Muscle insulin
resistance, due to ectopic lipid, precedes liver insulin resistance and diverts ingested
glucose to the liver, resulting in increased hepatic de novo lipogenesis and hyperlipidemia.
Subsequent macrophage infiltration into white adipose tissue (WAT) leads to increased
lipolysis, which further increases hepatic triglyceride synthesis and hyperlipidemia due to
increased fatty acid esterification. Macrophage-induced WAT lipolysis also stimulates
hepatic gluconeogenesis, promoting fasting and postprandial hyperglycemia through
increased fatty acid delivery to the liver, which results in increased hepatic acetyl-CoA
content, a potent activator of pyruvate carboxylase, and increased glycerol conversion to
glucose. These substrate-regulated processes are mostly independent of insulin signaling
in the liver but are dependent on insulin signaling in WAT, which becomes defective with
inflammation. Therapies that decrease ectopic lipid storage and diminish macrophage-
induced WAT lipolysis will reverse the root causes of type 2 diabetes.
Review
Find the latest version:
http://jci.me/77812/pdf

The Journal of Clinical InvestigationR e v i e w1 2jci.org Volume 126 N umber 1 J anuary 2016
In most natural habitats, calorie availability is scarce and unpre-
dictable, necessitating the evolution of systems for the efficient
storage and utilization of energy. But in our modern, mecha-
nized society, caloric demands are minimized, while highly pal-
atable, calorie-dense foods and beverages are readily available.
These changes have fostered the current pandemic of obesity and
comorbid conditions of nonalcoholic fatty liver disease (NAFLD),
atherosclerosis, and type 2 diabetes (T2D). Insulin resistance is a
common feature of all these diseases, and much effort has been
invested in delineating the pathogenesis of insulin resistance. We
will first review the role of insulin and nutrients (specifically glu-
cose and fatty acids) in nutrient storage (Figure 1) and then use
this framework to explore the various defects that give rise to insu-
lin resistance and T2D (Figure 2).
Postprandial hepatic glucose and lipid
metabolism
The simple act of eating rapidly shifts hepatic glucose metabolism
from glucose production to glucose storage, a complex transition
regulated by multiple factors including nutrients, alterations in
pancreatic and enteric hormones, and neural regulation. Insulin is
a crucial regulator of this transition, primarily by activating glyco-
gen synthase (1). The importance of insulin is seen in patients with
type 1 diabetes (T1D), who only synthesize one-third the amount
of hepatic glycogen as control subjects after a mixed meal (2). Yet,
hyperinsulinemia, in the absence of hyperglycemia, promotes
hepatic glycogen cycling with minimal net hepatic glycogen syn-
thesis (1). And hyperglycemia, without hyperinsulinemia, inhibits
hepatic glycogenolysis via glucose-mediated inhibition of glyco-
gen phosphorylase (3), with minimal net hepatic glycogen synthe-
sis (1). The combination of hyperinsulinemia and hyperglycemia
maximizes net hepatic glycogen synthesis (1). Other nutrients fur-
ther optimize net hepatic glycogen synthesis, such as activation of
glucokinase by catalytic quantities of fructose (4).
Hepatic insulin action requires a coordinated relay of intracel-
lular signals (Figure 1 and ref. 5). Insulin activates the insulin recep-
tor tyrosine kinase (IRTK), with subsequent activation of kinases
including 3-phosphoinositide-dependent kinase-1 (PDK1) and
mTORC2 (6), which converge on Akt phosphorylation (6–8). The
pattern of insulin delivery may also impact Akt phosphorylation,
with pulsatile portal delivery (which better mimics physiology)
leading to greater activation than continuous, fixed insulin deliv-
ery (9). Activation of Akt is the integral result of multiple inputs
to regulate hepatic glucose and lipid metabolism. This model has
been used to explain how insulin suppresses hepatic glucose pro-
duction via (i) lowering expression of gluconeogenic enzymes via
phosphorylation and nuclear exclusion of FOXO1 and (ii) inacti-
vation of glycogen synthase kinase 3β (GSK3β), which permits the
activation of glycogen synthase.
Recent studies challenge the primacy of the Akt/GSK3β/
glycogen synthase branch in the regulation of glycogen synthe-
sis (10). Hepatic insulin resistance in mice lacking both hepatic
Akt1 and Akt2 (double liver KO; DLKO) was essentially normal-
ized by the additional deletion of FOXO1, in a triple KO (TLKO)
mouse model (11). Remarkably, the TLKO mice exhibited rela-
tively normal fasting and postprandial glucose tolerance (10).
This suggests that FOXO1 deletion enables other mechanisms to
normalize hepatic glucose metabolism. Finally, Akt2-mediated
Insulin resistance arises when the nutrient storage pathways evolved to maximize efficient energy utilization are exposed
to chronic energy surplus. Ectopic lipid accumulation in liver and skeletal muscle triggers pathways that impair insulin
signaling, leading to reduced muscle glucose uptake and decreased hepatic glycogen synthesis. Muscle insulin resistance,
due to ectopic lipid, precedes liver insulin resistance and diverts ingested glucose to the liver, resulting in increased hepatic
de novo lipogenesis and hyperlipidemia. Subsequent macrophage infiltration into white adipose tissue (WAT) leads to
increased lipolysis, which further increases hepatic triglyceride synthesis and hyperlipidemia due to increased fatty
acid esterification. Macrophage-induced WAT lipolysis also stimulates hepatic gluconeogenesis, promoting fasting and
postprandial hyperglycemia through increased fatty acid delivery to the liver, which results in increased hepatic acetyl-
CoA content, a potent activator of pyruvate carboxylase, and increased glycerol conversion to glucose. These substrate-
regulated processes are mostly independent of insulin signaling in the liver but are dependent on insulin signaling in WAT,
which becomes defective with inflammation. Therapies that decrease ectopic lipid storage and diminish macrophage-
induced WAT lipolysis will reverse the root causes of type 2 diabetes.
The pathogenesis of insulin resistance: integrating
signaling pathways and substrate flux
Varman T. Samuel1,2 and Gerald I. Shulman 1,3,4
1
Department of Medicine, Yale University School of Medicine, New Haven, Connecticut, USA. 2Veterans Affairs Medical Center, West Haven, Connecticut, USA. 3 Department of Cellular and Molecular
Physiology and 4 Howard Hughes Medical Institute, Yale University School of Medicine, New Haven, Connecticut, USA.
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2016;126(1):12–22. doi:10.1172/JCI77812.
In most natural habitats, calorie availability is scarce and unpre-
dictable, necessitating the evolution of systems for the efficient
storage and utilization of energy. But in our modern, mecha-
nized society, caloric demands are minimized, while highly pal-
atable, calorie-dense foods and beverages are readily available.
These changes have fostered the current pandemic of obesity and
comorbid conditions of nonalcoholic fatty liver disease (NAFLD),
atherosclerosis, and type 2 diabetes (T2D). Insulin resistance is a
common feature of all these diseases, and much effort has been
invested in delineating the pathogenesis of insulin resistance. We
will first review the role of insulin and nutrients (specifically glu-
cose and fatty acids) in nutrient storage (Figure 1) and then use
this framework to explore the various defects that give rise to insu-
lin resistance and T2D (Figure 2).
Postprandial hepatic glucose and lipid
metabolism
The simple act of eating rapidly shifts hepatic glucose metabolism
from glucose production to glucose storage, a complex transition
regulated by multiple factors including nutrients, alterations in
pancreatic and enteric hormones, and neural regulation. Insulin is
a crucial regulator of this transition, primarily by activating glyco-
gen synthase (1). The importance of insulin is seen in patients with
type 1 diabetes (T1D), who only synthesize one-third the amount
of hepatic glycogen as control subjects after a mixed meal (2). Yet,
hyperinsulinemia, in the absence of hyperglycemia, promotes
hepatic glycogen cycling with minimal net hepatic glycogen syn-
thesis (1). And hyperglycemia, without hyperinsulinemia, inhibits
hepatic glycogenolysis via glucose-mediated inhibition of glyco-
gen phosphorylase (3), with minimal net hepatic glycogen synthe-
sis (1). The combination of hyperinsulinemia and hyperglycemia
maximizes net hepatic glycogen synthesis (1). Other nutrients fur-
ther optimize net hepatic glycogen synthesis, such as activation of
glucokinase by catalytic quantities of fructose (4).
Hepatic insulin action requires a coordinated relay of intracel-
lular signals (Figure 1 and ref. 5). Insulin activates the insulin recep-
tor tyrosine kinase (IRTK), with subsequent activation of kinases
including 3-phosphoinositide-dependent kinase-1 (PDK1) and
mTORC2 (6), which converge on Akt phosphorylation (6–8). The
pattern of insulin delivery may also impact Akt phosphorylation,
with pulsatile portal delivery (which better mimics physiology)
leading to greater activation than continuous, fixed insulin deliv-
ery (9). Activation of Akt is the integral result of multiple inputs
to regulate hepatic glucose and lipid metabolism. This model has
been used to explain how insulin suppresses hepatic glucose pro-
duction via (i) lowering expression of gluconeogenic enzymes via
phosphorylation and nuclear exclusion of FOXO1 and (ii) inacti-
vation of glycogen synthase kinase 3β (GSK3β), which permits the
activation of glycogen synthase.
Recent studies challenge the primacy of the Akt/GSK3β/
glycogen synthase branch in the regulation of glycogen synthe-
sis (10). Hepatic insulin resistance in mice lacking both hepatic
Akt1 and Akt2 (double liver KO; DLKO) was essentially normal-
ized by the additional deletion of FOXO1, in a triple KO (TLKO)
mouse model (11). Remarkably, the TLKO mice exhibited rela-
tively normal fasting and postprandial glucose tolerance (10).
This suggests that FOXO1 deletion enables other mechanisms to
normalize hepatic glucose metabolism. Finally, Akt2-mediated
Insulin resistance arises when the nutrient storage pathways evolved to maximize efficient energy utilization are exposed
to chronic energy surplus. Ectopic lipid accumulation in liver and skeletal muscle triggers pathways that impair insulin
signaling, leading to reduced muscle glucose uptake and decreased hepatic glycogen synthesis. Muscle insulin resistance,
due to ectopic lipid, precedes liver insulin resistance and diverts ingested glucose to the liver, resulting in increased hepatic
de novo lipogenesis and hyperlipidemia. Subsequent macrophage infiltration into white adipose tissue (WAT) leads to
increased lipolysis, which further increases hepatic triglyceride synthesis and hyperlipidemia due to increased fatty
acid esterification. Macrophage-induced WAT lipolysis also stimulates hepatic gluconeogenesis, promoting fasting and
postprandial hyperglycemia through increased fatty acid delivery to the liver, which results in increased hepatic acetyl-
CoA content, a potent activator of pyruvate carboxylase, and increased glycerol conversion to glucose. These substrate-
regulated processes are mostly independent of insulin signaling in the liver but are dependent on insulin signaling in WAT,
which becomes defective with inflammation. Therapies that decrease ectopic lipid storage and diminish macrophage-
induced WAT lipolysis will reverse the root causes of type 2 diabetes.
The pathogenesis of insulin resistance: integrating
signaling pathways and substrate flux
Varman T. Samuel1,2 and Gerald I. Shulman 1,3,4
1
Department of Medicine, Yale University School of Medicine, New Haven, Connecticut, USA. 2Veterans Affairs Medical Center, West Haven, Connecticut, USA. 3 Department of Cellular and Molecular
Physiology and 4 Howard Hughes Medical Institute, Yale University School of Medicine, New Haven, Connecticut, USA.
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2016;126(1):12–22. doi:10.1172/JCI77812.

The Journal of Clinical Investigation R e v i e w1 3jci.org Volume 126 N umber 1 J anuary 2016
suppression of hepatic glucose produc-
tion after meals (12, 13). In a canine study,
raising portal insulin eight-fold — while
keeping plasma glucose and glucagon
concentrations fixed — lowered hepatic
glucose production within 30 minutes but
did not reduce protein expression of glu-
coneogenic enzymes. Two hours of sus-
tained hyperinsulinemia were required
to detect a modest decline in protein (14).
More than 50 years ago, Levine and Fritz
proposed that insulin inhibited hepatic
glucose production through an indirect
mechanism (15). Bergman and others pos-
tulated that insulin’s ability to suppress
hepatic glucose production was linked to
the suppression of adipose lipolysis (16).
Recent studies by Perry et al. provide a
molecular mechanism linking insulin
action in white adipose tissue (WAT) to
the regulation of hepatic gluconeogenesis
(Figure 3). First, insulin suppressed adi-
pose lipolysis, lowered hepatic acetyl-CoA
content (an allosteric activator of pyruvate carboxylase [PC]),
and reduced PC activity and PC flux (17). Second, by inhibit-
ing lipolysis, insulin curtailed glycerol delivery to the liver and
reduced conversion of glycerol to glucose (18, 19). Hepatic insu-
lin signaling may establish the transcriptional tone of gluconeo-
genic enzymes and determine the gluconeogenic capacity of the
liver, but the ability of insulin to acutely regulate hepatic glu-
phosphorylation of GSK3 appears dispensable for normal hepatic
glucose metabolism (10). Thus, Akt activation is not a linchpin
for coordinating hepatic glucose metabolism, and other path-
ways also regulate this process.
Insulin-mediated suppression of hepatic glucose produc-
tion is often attributed to reduced transcription of gluconeo-
genic enzymes; however, this model fails to explain the rapid
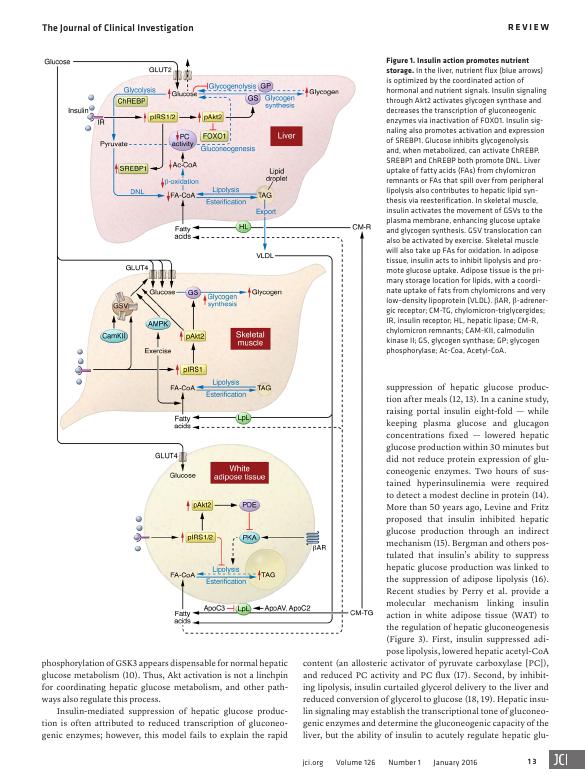
Figure 1. Insulin action promotes nutrient
storage. In the liver, nutrient flux (blue arrows)
is optimized by the coordinated action of
hormonal and nutrient signals. Insulin signaling
through Akt2 activates glycogen synthase and
decreases the transcription of gluconeogenic
enzymes via inactivation of FOXO1. Insulin sig-
naling also promotes activation and expression
of SREBP1. Glucose inhibits glycogenolysis
and, when metabolized, can activate ChREBP.
SREBP1 and ChREBP both promote DNL. Liver
uptake of fatty acids (FAs) from chylomicron
remnants or FAs that spill over from peripheral
lipolysis also contributes to hepatic lipid syn-
thesis via reesterification. In skeletal muscle,
insulin activates the movement of GSVs to the
plasma membrane, enhancing glucose uptake
and glycogen synthesis. GSV translocation can
also be activated by exercise. Skeletal muscle
will also take up FAs for oxidation. In adipose
tissue, insulin acts to inhibit lipolysis and pro-
mote glucose uptake. Adipose tissue is the pri-
mary storage location for lipids, with a coordi-
nate uptake of fats from chylomicrons and very
low–density lipoprotein (VLDL). βAR, β-adrener-
gic receptor; CM-TG, chylomicron-triglycergides;
IR, insulin receptor; HL, hepatic lipase; CM-R,
chylomicron remnants; CAM-KII, calmodulin
kinase II; GS, glycogen synthase; GP; glycogen
phosphorylase; Ac-Coa, Acetyl-CoA.
suppression of hepatic glucose produc-
tion after meals (12, 13). In a canine study,
raising portal insulin eight-fold — while
keeping plasma glucose and glucagon
concentrations fixed — lowered hepatic
glucose production within 30 minutes but
did not reduce protein expression of glu-
coneogenic enzymes. Two hours of sus-
tained hyperinsulinemia were required
to detect a modest decline in protein (14).
More than 50 years ago, Levine and Fritz
proposed that insulin inhibited hepatic
glucose production through an indirect
mechanism (15). Bergman and others pos-
tulated that insulin’s ability to suppress
hepatic glucose production was linked to
the suppression of adipose lipolysis (16).
Recent studies by Perry et al. provide a
molecular mechanism linking insulin
action in white adipose tissue (WAT) to
the regulation of hepatic gluconeogenesis
(Figure 3). First, insulin suppressed adi-
pose lipolysis, lowered hepatic acetyl-CoA
content (an allosteric activator of pyruvate carboxylase [PC]),
and reduced PC activity and PC flux (17). Second, by inhibit-
ing lipolysis, insulin curtailed glycerol delivery to the liver and
reduced conversion of glycerol to glucose (18, 19). Hepatic insu-
lin signaling may establish the transcriptional tone of gluconeo-
genic enzymes and determine the gluconeogenic capacity of the
liver, but the ability of insulin to acutely regulate hepatic glu-
phosphorylation of GSK3 appears dispensable for normal hepatic
glucose metabolism (10). Thus, Akt activation is not a linchpin
for coordinating hepatic glucose metabolism, and other path-
ways also regulate this process.
Insulin-mediated suppression of hepatic glucose produc-
tion is often attributed to reduced transcription of gluconeo-
genic enzymes; however, this model fails to explain the rapid
Figure 1. Insulin action promotes nutrient
storage. In the liver, nutrient flux (blue arrows)
is optimized by the coordinated action of
hormonal and nutrient signals. Insulin signaling
through Akt2 activates glycogen synthase and
decreases the transcription of gluconeogenic
enzymes via inactivation of FOXO1. Insulin sig-
naling also promotes activation and expression
of SREBP1. Glucose inhibits glycogenolysis
and, when metabolized, can activate ChREBP.
SREBP1 and ChREBP both promote DNL. Liver
uptake of fatty acids (FAs) from chylomicron
remnants or FAs that spill over from peripheral
lipolysis also contributes to hepatic lipid syn-
thesis via reesterification. In skeletal muscle,
insulin activates the movement of GSVs to the
plasma membrane, enhancing glucose uptake
and glycogen synthesis. GSV translocation can
also be activated by exercise. Skeletal muscle
will also take up FAs for oxidation. In adipose
tissue, insulin acts to inhibit lipolysis and pro-
mote glucose uptake. Adipose tissue is the pri-
mary storage location for lipids, with a coordi-
nate uptake of fats from chylomicrons and very
low–density lipoprotein (VLDL). βAR, β-adrener-
gic receptor; CM-TG, chylomicron-triglycergides;
IR, insulin receptor; HL, hepatic lipase; CM-R,
chylomicron remnants; CAM-KII, calmodulin
kinase II; GS, glycogen synthase; GP; glycogen
phosphorylase; Ac-Coa, Acetyl-CoA.
The Journal of Clinical InvestigationR e v i e w1 4jci.org Volume 126 N umber 1 J anuary 2016
skeletal muscle activates Akt2 (Figure 1).
Akt2 activation leads to phosphorylation
and inactivation of two RabGTPases,
Akt substrate of 160 kDa (AS160, also
known as TBC1D4; refs. 20, 21) and
TBC1D1 (22), which increase glucose
transporter type 4–containing (GLUT4-
containing) storage vesicles (GSVs) traf-
ficking to the plasma membrane, cellular
glucose transport, and glycogen synthe-
sis. Some recent studies also suggest the
presence of a PI3K-independent arm
that can promote cleavage of tether con-
taining UBX domain for GLUT4 (TUG),
a protein that sequesters GSVs in a per-
inuclear storage compartment (23–26).
Insulin-independent mechanisms also
activate muscle glucose uptake. Mice with
targeted deletion of the insulin receptor
in skeletal muscle have impaired insulin-
induced muscle glucose uptake (27) but
have normal exercise-induced muscle
glucose uptake (28). Muscle contraction
activates AMPK, which phosphorylates
proteins that regulate GSV translocation
(29). AMPK activation is one of many fac-
tors that comprise a network of signals
that promote glucose uptake in response
to exercise but independently of insulin
action (recently reviewed in ref. 30).
WAT glucose uptake is largely insulin dependent and regu-
lated by pathways similar to those in skeletal muscle (Figure 1).
WAT glucose uptake, quantitatively, is relatively minor,
accounting for only 5%–10% of whole body glucose uptake (31,
coneogenesis occurs mostly by an indirect mechanism through
inhibition of WAT lipolysis.
Muscle glycogen synthesis accounts for the majority of post-
prandial glucose disposal (14). As in the liver, insulin action in the
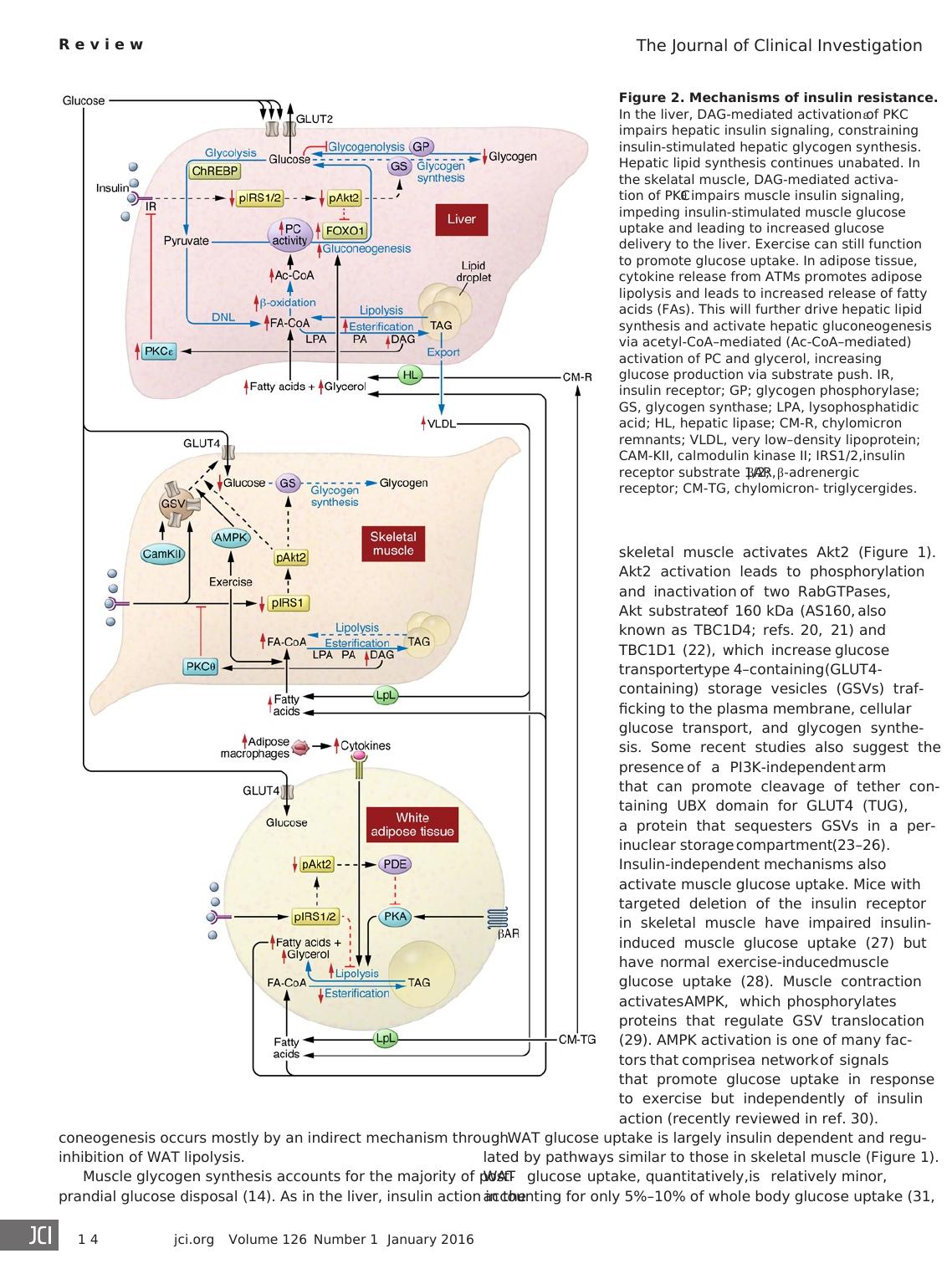
Figure 2. Mechanisms of insulin resistance.
In the liver, DAG-mediated activation of PKCε
impairs hepatic insulin signaling, constraining
insulin-stimulated hepatic glycogen synthesis.
Hepatic lipid synthesis continues unabated. In
the skelatal muscle, DAG-mediated activa-
tion of PKCθ impairs muscle insulin signaling,
impeding insulin-stimulated muscle glucose
uptake and leading to increased glucose
delivery to the liver. Exercise can still function
to promote glucose uptake. In adipose tissue,
cytokine release from ATMs promotes adipose
lipolysis and leads to increased release of fatty
acids (FAs). This will further drive hepatic lipid
synthesis and activate hepatic gluconeogenesis
via acetyl-CoA–mediated (Ac-CoA–mediated)
activation of PC and glycerol, increasing
glucose production via substrate push. IR,
insulin receptor; GP; glycogen phosphorylase;
GS, glycogen synthase; LPA, lysophosphatidic
acid; HL, hepatic lipase; CM-R, chylomicron
remnants; VLDL, very low–density lipoprotein;
CAM-KII, calmodulin kinase II; IRS1/2,insulin
receptor substrate 1/2; βAR, β-adrenergic
receptor; CM-TG, chylomicron- triglycergides.
skeletal muscle activates Akt2 (Figure 1).
Akt2 activation leads to phosphorylation
and inactivation of two RabGTPases,
Akt substrate of 160 kDa (AS160, also
known as TBC1D4; refs. 20, 21) and
TBC1D1 (22), which increase glucose
transporter type 4–containing (GLUT4-
containing) storage vesicles (GSVs) traf-
ficking to the plasma membrane, cellular
glucose transport, and glycogen synthe-
sis. Some recent studies also suggest the
presence of a PI3K-independent arm
that can promote cleavage of tether con-
taining UBX domain for GLUT4 (TUG),
a protein that sequesters GSVs in a per-
inuclear storage compartment (23–26).
Insulin-independent mechanisms also
activate muscle glucose uptake. Mice with
targeted deletion of the insulin receptor
in skeletal muscle have impaired insulin-
induced muscle glucose uptake (27) but
have normal exercise-induced muscle
glucose uptake (28). Muscle contraction
activates AMPK, which phosphorylates
proteins that regulate GSV translocation
(29). AMPK activation is one of many fac-
tors that comprise a network of signals
that promote glucose uptake in response
to exercise but independently of insulin
action (recently reviewed in ref. 30).
WAT glucose uptake is largely insulin dependent and regu-
lated by pathways similar to those in skeletal muscle (Figure 1).
WAT glucose uptake, quantitatively, is relatively minor,
accounting for only 5%–10% of whole body glucose uptake (31,
coneogenesis occurs mostly by an indirect mechanism through
inhibition of WAT lipolysis.
Muscle glycogen synthesis accounts for the majority of post-
prandial glucose disposal (14). As in the liver, insulin action in the
Figure 2. Mechanisms of insulin resistance.
In the liver, DAG-mediated activation of PKCε
impairs hepatic insulin signaling, constraining
insulin-stimulated hepatic glycogen synthesis.
Hepatic lipid synthesis continues unabated. In
the skelatal muscle, DAG-mediated activa-
tion of PKCθ impairs muscle insulin signaling,
impeding insulin-stimulated muscle glucose
uptake and leading to increased glucose
delivery to the liver. Exercise can still function
to promote glucose uptake. In adipose tissue,
cytokine release from ATMs promotes adipose
lipolysis and leads to increased release of fatty
acids (FAs). This will further drive hepatic lipid
synthesis and activate hepatic gluconeogenesis
via acetyl-CoA–mediated (Ac-CoA–mediated)
activation of PC and glycerol, increasing
glucose production via substrate push. IR,
insulin receptor; GP; glycogen phosphorylase;
GS, glycogen synthase; LPA, lysophosphatidic
acid; HL, hepatic lipase; CM-R, chylomicron
remnants; VLDL, very low–density lipoprotein;
CAM-KII, calmodulin kinase II; IRS1/2,insulin
receptor substrate 1/2; βAR, β-adrenergic
receptor; CM-TG, chylomicron- triglycergides.
End of preview
Want to access all the pages? Upload your documents or become a member.
Related Documents
Biochemistry Assignment (Doc)lg...
|5
|921
|119
Report on Diabetes Mellitus Medicinelg...
|5
|854
|219