Report on Protein Structure, Function, and Prediction Methods
VerifiedAdded on 2022/11/16
|10
|2711
|447
Report
AI Summary
This report provides a comprehensive overview of protein structure, beginning with the building blocks of proteins, amino acids, and their classification. It then delves into the different levels of protein structure: primary, secondary (alpha-helices and beta-sheets), tertiary, and quaternary structures, explaining the forces that stabilize each level. The report discusses the role of torsion angles and the Ramachandran plot in understanding protein conformations. Furthermore, it explores the application of machine learning in protein structure prediction, highlighting the algorithms and techniques used to predict protein structure from amino acid sequences and the potential of machine learning in this field. The report also touches upon the limitations and future directions of machine learning in computational biophysics.

Proteins
Proteins are polymers of amino acids [1]. In nature, 20 amino acids exist that arrange in
different linear combinations through condensation by formation of a peptide bond to give
rise to short sequences known as peptides. These peptides through polymerization and
formation of covalent bonds ultimately form proteins. In a cell, proteins form structural
components, participate in signalling cascades, perform cell recognition and adhesion and
also function as biocatalysts in biochemical reactions [2].
Amino Acids
Amino acids are monomers of proteins [3]. In nature, so far, 20 amino acids are known to
exist that participate in various cellular reactions and in the formation of proteins. All amino
acids contain a central carbon atom (C) which has a valency of four [4]. It is linked to an
amino group (NH2) on one side and to a carboxyl group (COOH) on the other. The central
carbon, also known as the alpha (α) carbon is linked to a hydrogen atom (H) and to a side
chain (R). Among the different amino acids, the R group differs while the other three groups
remain conserved [5]. The general structure of an amino acid is shown below:
Figure 1: General structure of an amino acid
Since the amino group and the carboxyl group are linked to the central alpha carbon atom,
these amino acids are referred to as the α-amino acids. The 20 α -amino acids possess
different physical and chemical properties that are imparted to them by the virtue of the R
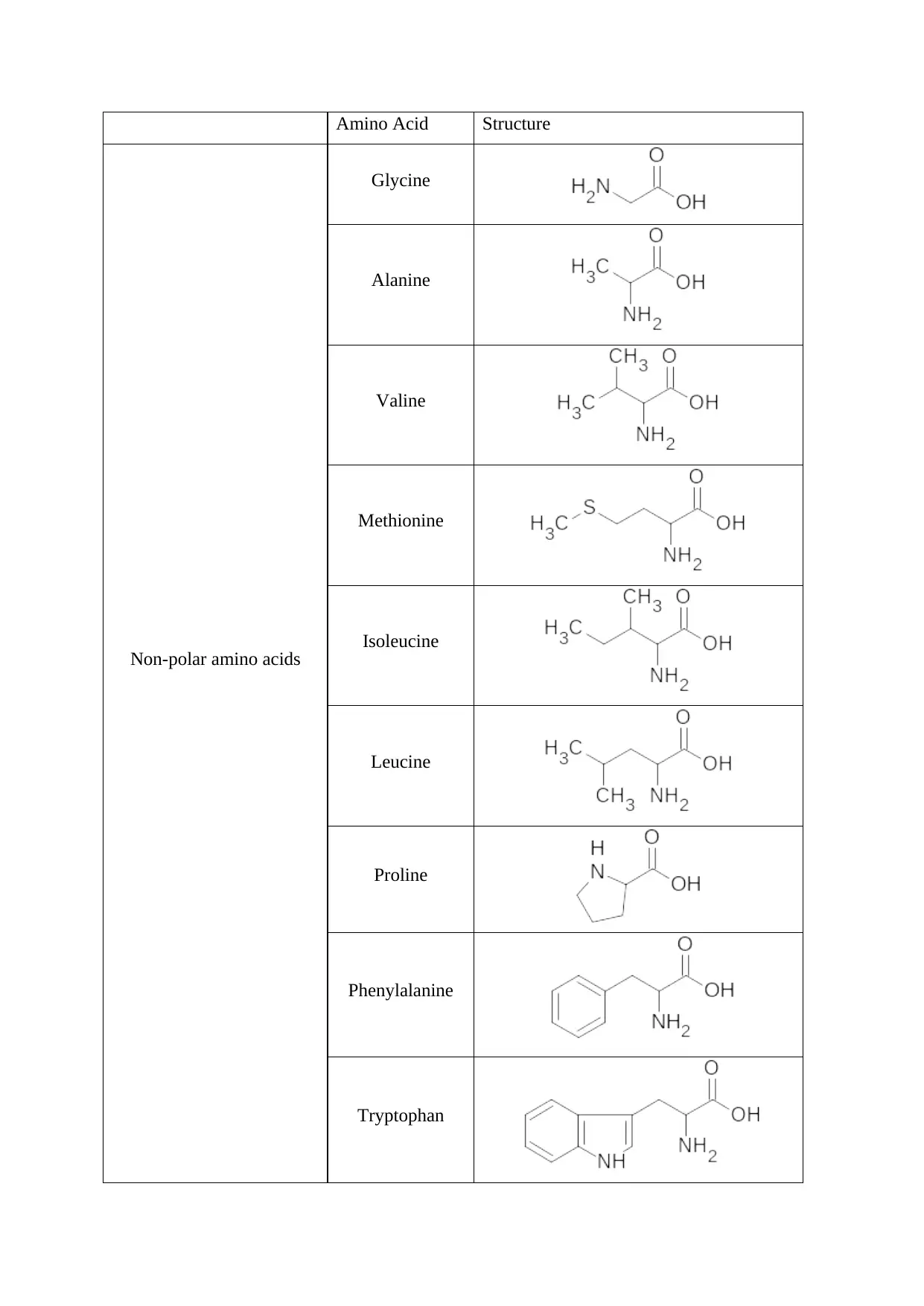
group. The twenty α-amino acids are classified into different groups, such as- polar, non-
polar, acidic, basic amino acids, on the basis of the differences in the R group [6]. Some
prominent examples from different subs-classes of amino acids are:
Proteins are polymers of amino acids [1]. In nature, 20 amino acids exist that arrange in
different linear combinations through condensation by formation of a peptide bond to give
rise to short sequences known as peptides. These peptides through polymerization and
formation of covalent bonds ultimately form proteins. In a cell, proteins form structural
components, participate in signalling cascades, perform cell recognition and adhesion and
also function as biocatalysts in biochemical reactions [2].
Amino Acids
Amino acids are monomers of proteins [3]. In nature, so far, 20 amino acids are known to
exist that participate in various cellular reactions and in the formation of proteins. All amino
acids contain a central carbon atom (C) which has a valency of four [4]. It is linked to an
amino group (NH2) on one side and to a carboxyl group (COOH) on the other. The central
carbon, also known as the alpha (α) carbon is linked to a hydrogen atom (H) and to a side
chain (R). Among the different amino acids, the R group differs while the other three groups
remain conserved [5]. The general structure of an amino acid is shown below:
Figure 1: General structure of an amino acid
Since the amino group and the carboxyl group are linked to the central alpha carbon atom,
these amino acids are referred to as the α-amino acids. The 20 α -amino acids possess
different physical and chemical properties that are imparted to them by the virtue of the R
group. The twenty α-amino acids are classified into different groups, such as- polar, non-
polar, acidic, basic amino acids, on the basis of the differences in the R group [6]. Some
prominent examples from different subs-classes of amino acids are:
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser
Amino Acid Structure
Non-polar amino acids
Glycine
Alanine
Valine
Methionine
Isoleucine
Leucine
Proline
Phenylalanine
Tryptophan
Non-polar amino acids
Glycine
Alanine
Valine
Methionine
Isoleucine
Leucine
Proline
Phenylalanine
Tryptophan
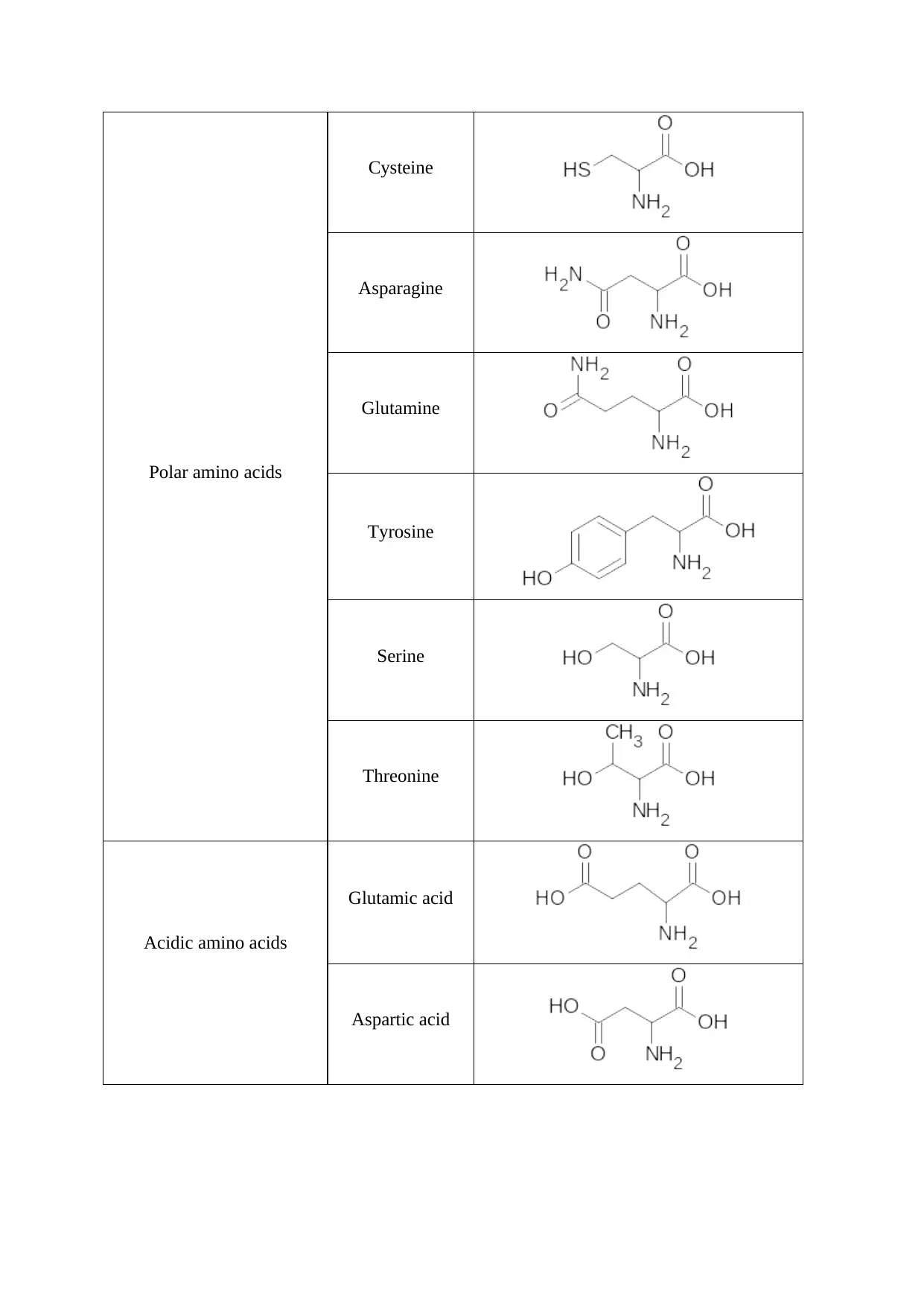
Polar amino acids
Cysteine
Asparagine
Glutamine
Tyrosine
Serine
Threonine
Acidic amino acids
Glutamic acid
Aspartic acid
Cysteine
Asparagine
Glutamine
Tyrosine
Serine
Threonine
Acidic amino acids
Glutamic acid
Aspartic acid
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

Basic amino Acids
Histidine
Lysine
Arginine
The hydrophobic R groups make an amino acid non-polar. Such amino acid residues tend to
aggregate together in an aqueous environment. While polar R groups are present in
hydrophilic amino acids and participate in hydrogen bonding and other hydrophilic
interactions. The acidic amino acids possess a carboxylate group that bears a negative charge
at physiological pH. Such amino acids form active residues of enzymes and participate in
biochemical reactions. Amino acids with basic side chains are often involved in enzyme
catalysed reactions and hydrogen bonding.
Protein Structure: Overview
In biological systems, proteins perform a variety of important functions [7]. There have been
various reports to establish a relation between protein function and their structure [8].
Further, protein structure is determined by the sequence of amino acids in the protein chain
[9]. Due to various interactive forces that exist among the constituent amino acids and the
side chains, a protein tends to occupy an energetically favourable structure. Protein structure
is best understood at four levels- primary, secondary, tertiary and quaternary structures [10].
Primary structure
The primary structure is represented by the sequence of amino acids in the protein chain, also
known as the polypeptide [11]. The various constituent amino acids are linked together by
peptide bonds that are formed by condensation of amino group on α-carbon of one amino
Histidine
Lysine
Arginine
The hydrophobic R groups make an amino acid non-polar. Such amino acid residues tend to
aggregate together in an aqueous environment. While polar R groups are present in
hydrophilic amino acids and participate in hydrogen bonding and other hydrophilic
interactions. The acidic amino acids possess a carboxylate group that bears a negative charge
at physiological pH. Such amino acids form active residues of enzymes and participate in
biochemical reactions. Amino acids with basic side chains are often involved in enzyme
catalysed reactions and hydrogen bonding.
Protein Structure: Overview
In biological systems, proteins perform a variety of important functions [7]. There have been
various reports to establish a relation between protein function and their structure [8].
Further, protein structure is determined by the sequence of amino acids in the protein chain
[9]. Due to various interactive forces that exist among the constituent amino acids and the
side chains, a protein tends to occupy an energetically favourable structure. Protein structure
is best understood at four levels- primary, secondary, tertiary and quaternary structures [10].
Primary structure
The primary structure is represented by the sequence of amino acids in the protein chain, also
known as the polypeptide [11]. The various constituent amino acids are linked together by
peptide bonds that are formed by condensation of amino group on α-carbon of one amino
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

acid with the carboxyl group on the α-carbon of the other. This is the simplest level of protein
structure in which the protein is represented as a linear sequence of its amino acids
Secondary structure
The secondary structure of proteins arises due to hydrogen bonding between amino and
carboxyl groups of amino acids [12]. Two common types of secondary structures that are
known so far are- the α- helices and the β- sheets.
α – helix
It is a helical structure that arises due to the formation of H-bond between the N-H group and
the oxygen of a C=O group, at a difference of four amino acids in the next turn of the helix. A
typical α- helix is about eleven amino acids long. A helix can be right handed or left handed
in direction. The stability of an α-helix is dependent on steric interactions between the amino
acids. The presence of amino acids with bulky R-groups (for example- tryptophan, tyrosine)
or too small R groups (like glycine) tends to make α-helices less stable. Proline, which is
characterized by its irregular geometry arising due to its R-group which bonds back to the
nitrogen of the amide group resulting in steric hindrance, also destabilizes α-helices.
Moreover, proline lacks hydrogen on its nitrogen atom. This prevents proline from
participating in hydrogen bonding which ultimately affects the helical structure.
β- sheet
Also known as the pleated β-sheet, consists of laterally attached proteins strands that are held
together by H-bonds. Here the hydrogen bonds are formed between amine and carbonyl
groups of two protein chains rather than within the protein chain.
Tertiary structure
This level of protein structure refers to the three dimensional shape of the proteins. It is
formed as a result of folding and twisting of the secondary structures in a 3-D space. The
tertiary structure is stabilized by hydrogen bonds, hydrophobic and hydrophilic interactions,
ionic bonds and disulphide bonds. Tertiary structure of proteins categorised into two main
types- globular and fibrous.
Quaternary Structure
It refers to the protein structure arising due the interactions between two or more polypeptide
chains that are held together by covalent bonds or through an organic component known as
structure in which the protein is represented as a linear sequence of its amino acids
Secondary structure
The secondary structure of proteins arises due to hydrogen bonding between amino and
carboxyl groups of amino acids [12]. Two common types of secondary structures that are
known so far are- the α- helices and the β- sheets.
α – helix
It is a helical structure that arises due to the formation of H-bond between the N-H group and
the oxygen of a C=O group, at a difference of four amino acids in the next turn of the helix. A
typical α- helix is about eleven amino acids long. A helix can be right handed or left handed
in direction. The stability of an α-helix is dependent on steric interactions between the amino
acids. The presence of amino acids with bulky R-groups (for example- tryptophan, tyrosine)
or too small R groups (like glycine) tends to make α-helices less stable. Proline, which is
characterized by its irregular geometry arising due to its R-group which bonds back to the
nitrogen of the amide group resulting in steric hindrance, also destabilizes α-helices.
Moreover, proline lacks hydrogen on its nitrogen atom. This prevents proline from
participating in hydrogen bonding which ultimately affects the helical structure.
β- sheet
Also known as the pleated β-sheet, consists of laterally attached proteins strands that are held
together by H-bonds. Here the hydrogen bonds are formed between amine and carbonyl
groups of two protein chains rather than within the protein chain.
Tertiary structure
This level of protein structure refers to the three dimensional shape of the proteins. It is
formed as a result of folding and twisting of the secondary structures in a 3-D space. The
tertiary structure is stabilized by hydrogen bonds, hydrophobic and hydrophilic interactions,
ionic bonds and disulphide bonds. Tertiary structure of proteins categorised into two main
types- globular and fibrous.
Quaternary Structure
It refers to the protein structure arising due the interactions between two or more polypeptide
chains that are held together by covalent bonds or through an organic component known as

the prosthetic group. Quaternary structure is often categorised on the basis of subunits
involved- homodimer (if all the subunits are identical) or heterodimer (if the subunits are
different). Common examples include- haemoglobin and collagen.
Secondary structure - torsion angles
The peptide bond in proteins results from the sharing of electrons between C, N and O atoms.
Since the electronegativity difference of these atoms is small, the electron cloud tends to be
delocalized. This phenomenon imparts a partial double bond character to the peptide bond.
As a result, the rotation of this bond is hindered. However the other two bonds, namely the
Cα-C and the Cα-N, are free to rotate along the bond axis as shown below:
Also known as the dihedral angle, torsion angles are formed by three consecutive bonds in a
polypetide and are defined by the angle created between the two outer bonds. The rotation
along the Cα-C axis give rise to psi (ψ) torsion angle while rotation along the Cα-N results in
phi (φ) torsion angle. The limited rotation of the peptide bond (C-N) is represented by the
Omega (ω) torsion angle. For the twenty side chains in found in the amino acids that
contribute to protein formation, encoded are acidic, some basic, neutral, hydrophobic or
hydrophilic. These side chains contribute to steric hindrances and limit the torsion angles that
can exist for a given structure of protein. The rotation of torsion angles is best represented by
the Ramachandran plot.
Ramachandran plot [13]
The Ramachandran plot is a 2-D representation of the rotation of the torsion angles in of a
polypeptide and serves as a simple representation of the conformation of a protein. The φ and
ψ angle values are clustered into distinct regions in the Ramachandran plot on the basis of
steric clashes. Atoms are assumed as hard spheres with dimensions corresponding to their van
der Waals radius and the φ and ψ angle values resulting in collision of these spheres are
regarded as sterically disallowed conformations.
involved- homodimer (if all the subunits are identical) or heterodimer (if the subunits are
different). Common examples include- haemoglobin and collagen.
Secondary structure - torsion angles
The peptide bond in proteins results from the sharing of electrons between C, N and O atoms.
Since the electronegativity difference of these atoms is small, the electron cloud tends to be
delocalized. This phenomenon imparts a partial double bond character to the peptide bond.
As a result, the rotation of this bond is hindered. However the other two bonds, namely the
Cα-C and the Cα-N, are free to rotate along the bond axis as shown below:
Also known as the dihedral angle, torsion angles are formed by three consecutive bonds in a
polypetide and are defined by the angle created between the two outer bonds. The rotation
along the Cα-C axis give rise to psi (ψ) torsion angle while rotation along the Cα-N results in
phi (φ) torsion angle. The limited rotation of the peptide bond (C-N) is represented by the
Omega (ω) torsion angle. For the twenty side chains in found in the amino acids that
contribute to protein formation, encoded are acidic, some basic, neutral, hydrophobic or
hydrophilic. These side chains contribute to steric hindrances and limit the torsion angles that
can exist for a given structure of protein. The rotation of torsion angles is best represented by
the Ramachandran plot.
Ramachandran plot [13]
The Ramachandran plot is a 2-D representation of the rotation of the torsion angles in of a
polypeptide and serves as a simple representation of the conformation of a protein. The φ and
ψ angle values are clustered into distinct regions in the Ramachandran plot on the basis of
steric clashes. Atoms are assumed as hard spheres with dimensions corresponding to their van
der Waals radius and the φ and ψ angle values resulting in collision of these spheres are
regarded as sterically disallowed conformations.
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide
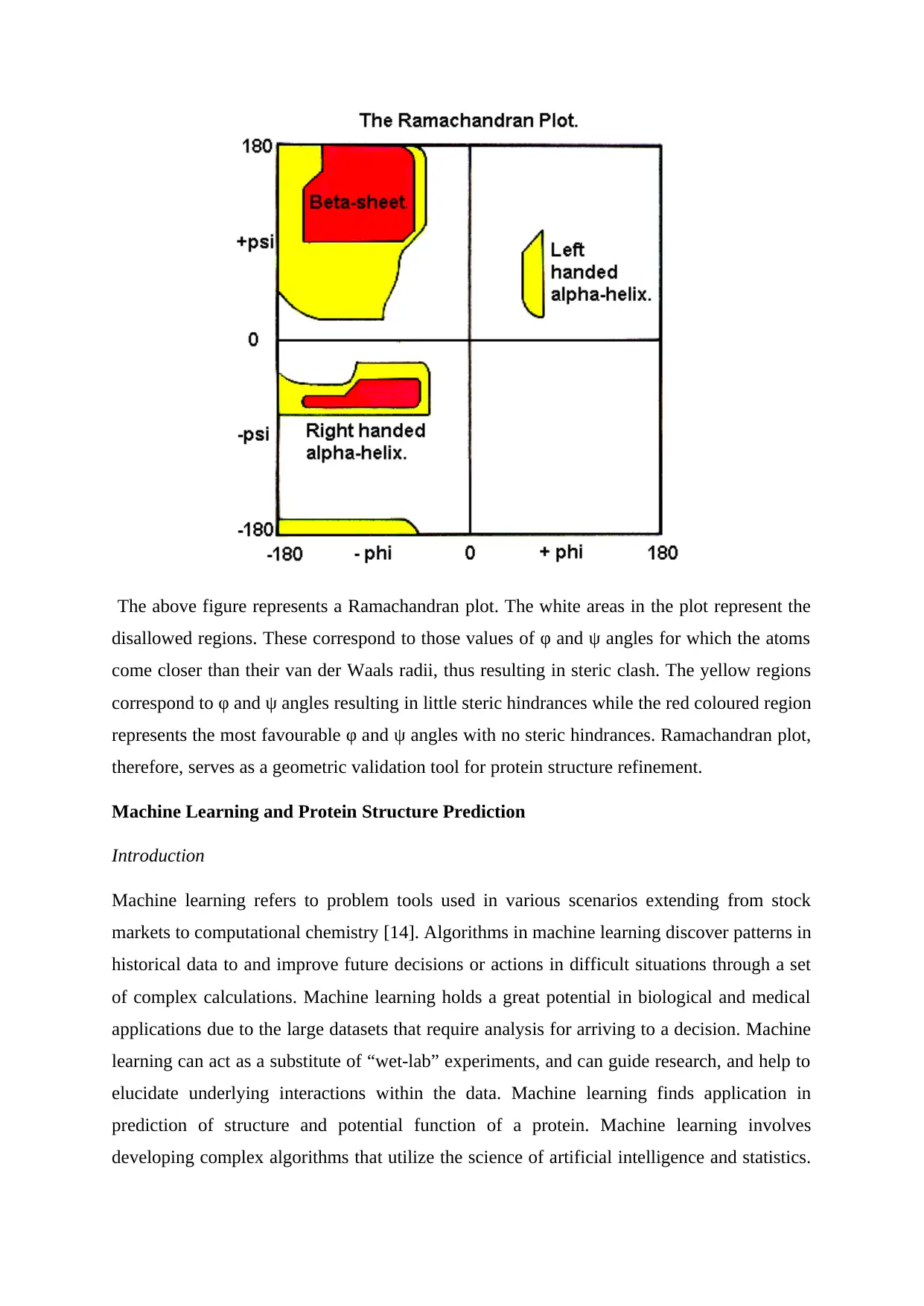
The above figure represents a Ramachandran plot. The white areas in the plot represent the
disallowed regions. These correspond to those values of φ and ψ angles for which the atoms
come closer than their van der Waals radii, thus resulting in steric clash. The yellow regions
correspond to φ and ψ angles resulting in little steric hindrances while the red coloured region
represents the most favourable φ and ψ angles with no steric hindrances. Ramachandran plot,
therefore, serves as a geometric validation tool for protein structure refinement.
Machine Learning and Protein Structure Prediction
Introduction
Machine learning refers to problem tools used in various scenarios extending from stock
markets to computational chemistry [14]. Algorithms in machine learning discover patterns in
historical data to and improve future decisions or actions in difficult situations through a set
of complex calculations. Machine learning holds a great potential in biological and medical
applications due to the large datasets that require analysis for arriving to a decision. Machine
learning can act as a substitute of “wet-lab” experiments, and can guide research, and help to
elucidate underlying interactions within the data. Machine learning finds application in
prediction of structure and potential function of a protein. Machine learning involves
developing complex algorithms that utilize the science of artificial intelligence and statistics.
disallowed regions. These correspond to those values of φ and ψ angles for which the atoms
come closer than their van der Waals radii, thus resulting in steric clash. The yellow regions
correspond to φ and ψ angles resulting in little steric hindrances while the red coloured region
represents the most favourable φ and ψ angles with no steric hindrances. Ramachandran plot,
therefore, serves as a geometric validation tool for protein structure refinement.
Machine Learning and Protein Structure Prediction
Introduction
Machine learning refers to problem tools used in various scenarios extending from stock
markets to computational chemistry [14]. Algorithms in machine learning discover patterns in
historical data to and improve future decisions or actions in difficult situations through a set
of complex calculations. Machine learning holds a great potential in biological and medical
applications due to the large datasets that require analysis for arriving to a decision. Machine
learning can act as a substitute of “wet-lab” experiments, and can guide research, and help to
elucidate underlying interactions within the data. Machine learning finds application in
prediction of structure and potential function of a protein. Machine learning involves
developing complex algorithms that utilize the science of artificial intelligence and statistics.
Paraphrase This Document
Need a fresh take? Get an instant paraphrase of this document with our AI Paraphraser

Some common examples of algorithms used in machine learning include neural networks and
decision trees and have been in use for many years and are still being used. At present there
are two basic types of machine learning algorithms: frequentist and evidential. In biology,
machine learning has gained importance and popularity due to the demand for the analysis of
high throughput data arising out of microarray analysis and sequencing. In recent years, the
potential of machine learning for protein structure prediction and function determination has
also been explored.
Protein structure prediction
Prediction of secondary structure of proteins from the primary structure has been a long
studied problem in biology [15]. Many of the successful method in current times employ
evolutionary information and rely on iterative search tools such as PSI-BLAST. The
prediction of secondary structure requires using many approaches such as neural or Bayesian
networks and others. In addition, structured prediction is required as the structure of a protein
depends on the interactions of a particular residue with other neighbours. Some of the
successful methods use two stage approach in which the first creates a multiclass prediction
and the second evaluates the best assignment by applying the information from the first stage.
Prediction of protein structure occupies a key position in understanding protein function [16].
The information about a protein’s structure enables to unlock a set of important features that
can be used to understand the protein function. Since experimental methods, like x-ray
crystallography and nuclear magnetic resonance (NMR) spectroscopy, expensive and are
time consuming, machine learning based structure prediction can play an important part in
attending to the issue. Further, since there is large a number of protein structures in the PDB,
the number of sequences for which structures are yet to be solved, are far greater. CATH and
SCOP are two systems that are used to label training data for a number of supervised learning
problems found in protein structure prediction and organization.
Several techniques have been successfully developed to determine sequence similarity. Since
sequences are known to dictate the structure in proteins, sequences that have similarity tend
to have the same structure. On the other hand, when the sequence similarity is less than a
threshold, the sequence homology based modelling fails to yield reliable results. Under such
circumstances, machine learning approach can play a key role to search for a template
sequence with known structure. One of the approaches utilizes efficient representation of
kernel classifiers while the other, known as the semi-supervised learning, utilizes the large
decision trees and have been in use for many years and are still being used. At present there
are two basic types of machine learning algorithms: frequentist and evidential. In biology,
machine learning has gained importance and popularity due to the demand for the analysis of
high throughput data arising out of microarray analysis and sequencing. In recent years, the
potential of machine learning for protein structure prediction and function determination has
also been explored.
Protein structure prediction
Prediction of secondary structure of proteins from the primary structure has been a long
studied problem in biology [15]. Many of the successful method in current times employ
evolutionary information and rely on iterative search tools such as PSI-BLAST. The
prediction of secondary structure requires using many approaches such as neural or Bayesian
networks and others. In addition, structured prediction is required as the structure of a protein
depends on the interactions of a particular residue with other neighbours. Some of the
successful methods use two stage approach in which the first creates a multiclass prediction
and the second evaluates the best assignment by applying the information from the first stage.
Prediction of protein structure occupies a key position in understanding protein function [16].
The information about a protein’s structure enables to unlock a set of important features that
can be used to understand the protein function. Since experimental methods, like x-ray
crystallography and nuclear magnetic resonance (NMR) spectroscopy, expensive and are
time consuming, machine learning based structure prediction can play an important part in
attending to the issue. Further, since there is large a number of protein structures in the PDB,
the number of sequences for which structures are yet to be solved, are far greater. CATH and
SCOP are two systems that are used to label training data for a number of supervised learning
problems found in protein structure prediction and organization.
Several techniques have been successfully developed to determine sequence similarity. Since
sequences are known to dictate the structure in proteins, sequences that have similarity tend
to have the same structure. On the other hand, when the sequence similarity is less than a
threshold, the sequence homology based modelling fails to yield reliable results. Under such
circumstances, machine learning approach can play a key role to search for a template
sequence with known structure. One of the approaches utilizes efficient representation of
kernel classifiers while the other, known as the semi-supervised learning, utilizes the large

amount unlabelled sequence data to build models similar to PSI-Blast. In addition to these,
some other methods utilise sequence-structure correlations or motifs in addition to kernel
methods.
Machine learning has become indispensable in protein structure prediction. In recent times,
machine learning has shown promising applications in protein structure and function
prediction, however it is still in nascent stages and a lot has to be developed for its use as one
of the main tools for computational biophysics. Most of the previous works relied on direct
application of available machine learning software in conjunction with simple features.
Further progress in this field requires breakthroughs beyond the straightforward application
of classification software packages. Machine learning represents a powerful tool and has
great potential applications in biomolecular modelling. In combination with biochemical and
biophysical information, it is expected to yield greater advancement in the understanding of
protein structures, functions, interactions and localizations.
References
[1]J. Berg, J. Tymoczko and L. Stryer, "Biochemistry", Ncbi.nlm.nih.gov, 2019. [Online].
Available: https://www.ncbi.nlm.nih.gov/books/NBK21154/.
[2]G. Cooper, "The Cell", Ncbi.nlm.nih.gov, 2019. [Online]. Available:
https://www.ncbi.nlm.nih.gov/books/NBK9839/. [Accessed: 26- May- 2019].
[3]"Chemistry for Biologists: Proteins", Rsc.org, 2019. [Online]. Available:
https://www.rsc.org/Education/Teachers/Resources/cfb/proteins.htm. [Accessed: 26- May-
2019].
[4]B. Kuhlman and D. Baker, "Native protein sequences are close to optimal for their
structures", Proceedings of the National Academy of Sciences, vol. 97, no. 19, pp. 10383-
10388, 2000. Available: 10.1073/pnas.97.19.10383 [Accessed 26 May 2019].
[5]B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, "Molecular Biology
of the Cell", Ncbi.nlm.nih.gov, 2019. [Online]. Available:
https://www.ncbi.nlm.nih.gov/books/NBK21054/. [Accessed: 26- May- 2019].
[6]O. Sparkman, Z. Penton and F. Kitson, Gas chromatography and mass spectrometry.
Burlington, MA: Academic Press, 2011.
[7]J. Berg, J. Tymoczko and L. Stryer, "Biochemistry", Ncbi.nlm.nih.gov, 2019. [Online].
Available: https://www.ncbi.nlm.nih.gov/books/NBK21154/. [Accessed: 26- May- 2019].
[8]G. Petsko and D. Ringe, Protein structure and function. Oxford [England]: Oxford
University Press, 2009.
[9]D. Whitford, Protein structure & function. New York: Wiley, 2003.
[10]M. Ryadnov and F. Hudecz, Amino Acids, Peptides and Proteins 42. [S.l.]: Royal Society
of Chemistry, 2018.
some other methods utilise sequence-structure correlations or motifs in addition to kernel
methods.
Machine learning has become indispensable in protein structure prediction. In recent times,
machine learning has shown promising applications in protein structure and function
prediction, however it is still in nascent stages and a lot has to be developed for its use as one
of the main tools for computational biophysics. Most of the previous works relied on direct
application of available machine learning software in conjunction with simple features.
Further progress in this field requires breakthroughs beyond the straightforward application
of classification software packages. Machine learning represents a powerful tool and has
great potential applications in biomolecular modelling. In combination with biochemical and
biophysical information, it is expected to yield greater advancement in the understanding of
protein structures, functions, interactions and localizations.
References
[1]J. Berg, J. Tymoczko and L. Stryer, "Biochemistry", Ncbi.nlm.nih.gov, 2019. [Online].
Available: https://www.ncbi.nlm.nih.gov/books/NBK21154/.
[2]G. Cooper, "The Cell", Ncbi.nlm.nih.gov, 2019. [Online]. Available:
https://www.ncbi.nlm.nih.gov/books/NBK9839/. [Accessed: 26- May- 2019].
[3]"Chemistry for Biologists: Proteins", Rsc.org, 2019. [Online]. Available:
https://www.rsc.org/Education/Teachers/Resources/cfb/proteins.htm. [Accessed: 26- May-
2019].
[4]B. Kuhlman and D. Baker, "Native protein sequences are close to optimal for their
structures", Proceedings of the National Academy of Sciences, vol. 97, no. 19, pp. 10383-
10388, 2000. Available: 10.1073/pnas.97.19.10383 [Accessed 26 May 2019].
[5]B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, "Molecular Biology
of the Cell", Ncbi.nlm.nih.gov, 2019. [Online]. Available:
https://www.ncbi.nlm.nih.gov/books/NBK21054/. [Accessed: 26- May- 2019].
[6]O. Sparkman, Z. Penton and F. Kitson, Gas chromatography and mass spectrometry.
Burlington, MA: Academic Press, 2011.
[7]J. Berg, J. Tymoczko and L. Stryer, "Biochemistry", Ncbi.nlm.nih.gov, 2019. [Online].
Available: https://www.ncbi.nlm.nih.gov/books/NBK21154/. [Accessed: 26- May- 2019].
[8]G. Petsko and D. Ringe, Protein structure and function. Oxford [England]: Oxford
University Press, 2009.
[9]D. Whitford, Protein structure & function. New York: Wiley, 2003.
[10]M. Ryadnov and F. Hudecz, Amino Acids, Peptides and Proteins 42. [S.l.]: Royal Society
of Chemistry, 2018.
⊘ This is a preview!⊘
Do you want full access?
Subscribe today to unlock all pages.
Trusted by 1+ million students worldwide

[11]C. Branden and J. Tooze, Introduction to Protein Structure. 2012.
[12]D. Chasman, Protein structure. New York: Dekker, 2003.
[13] J. Russell and R. Cohn, Ramachandran plot. [Place of publication not identified]: Book
On Demand, 2012.
[14]H. Bock, W. Lenski and M. Richter, Information Systems and Data Analysis. Berlin,
Heidelberg: Springer Berlin Heidelberg, 1994.
[15]Y. Zhou, A. Kloczkowski, E. Faraggi and Y. Yang, Prediction of protein secondary
structure.
[16]R. Huzefa and G. Karypis, Introduction to protein structure prediction. 2010.
[12]D. Chasman, Protein structure. New York: Dekker, 2003.
[13] J. Russell and R. Cohn, Ramachandran plot. [Place of publication not identified]: Book
On Demand, 2012.
[14]H. Bock, W. Lenski and M. Richter, Information Systems and Data Analysis. Berlin,
Heidelberg: Springer Berlin Heidelberg, 1994.
[15]Y. Zhou, A. Kloczkowski, E. Faraggi and Y. Yang, Prediction of protein secondary
structure.
[16]R. Huzefa and G. Karypis, Introduction to protein structure prediction. 2010.
1 out of 10
Related Documents






Your All-in-One AI-Powered Toolkit for Academic Success.
+13062052269
info@desklib.com
Available 24*7 on WhatsApp / Email
![[object Object]](/_next/static/media/star-bottom.7253800d.svg)
Unlock your academic potential
Copyright © 2020–2026 A2Z Services. All Rights Reserved. Developed and managed by ZUCOL.